Summary
Experimental structure of proteins collected using cryo-EM and X-ray-crystallography are higher-quality in ligand-binding areas than other areas (1).
Figures
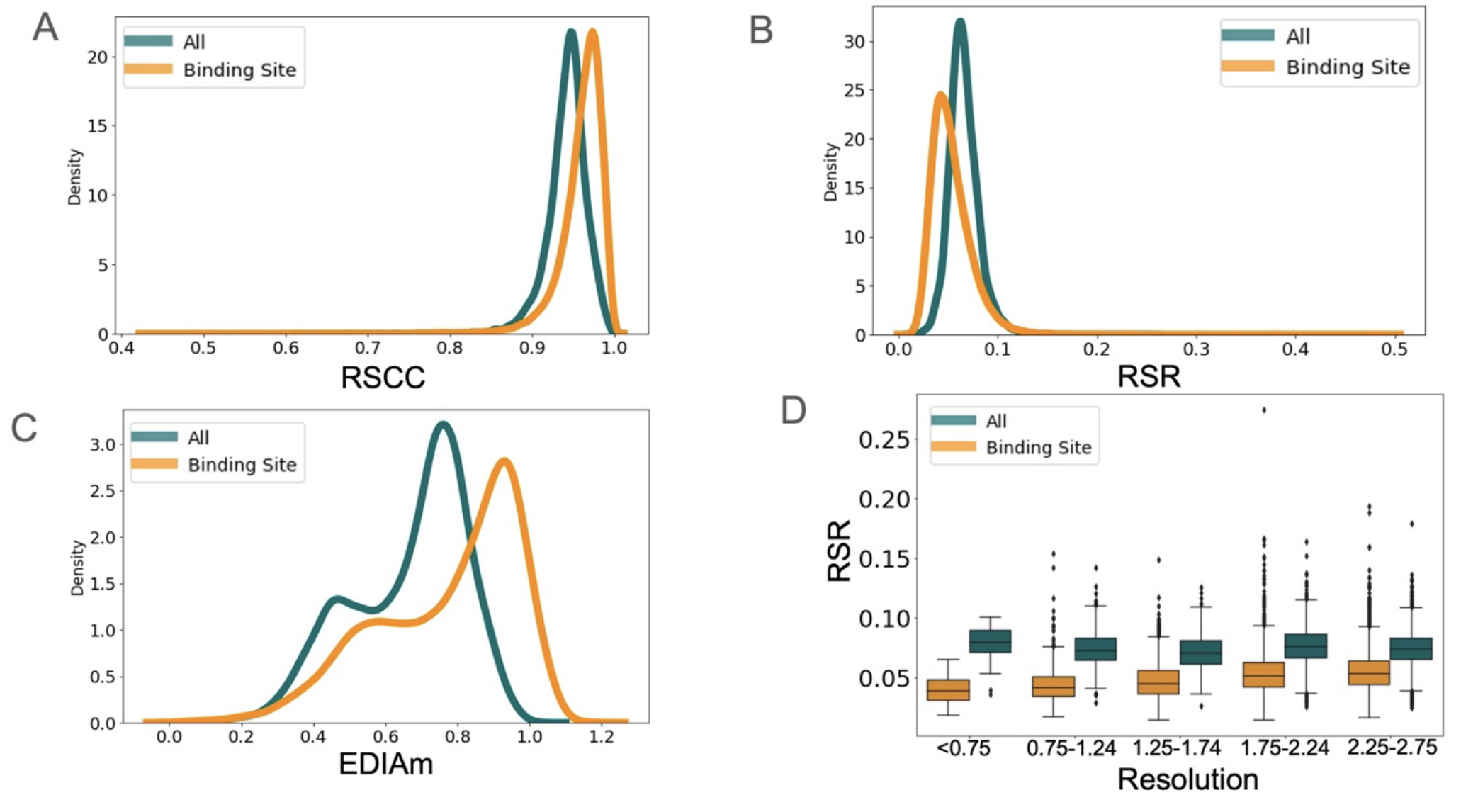 Ref (1)
Ref (1)
1.
Wankowicz SA. Modeling Bias Toward Binding Sites in PDB Structural Models. openRxiv; 2024. Available from: https://doi.org/10.1101/2024.12.14.628518