Free energy perturbation (FEP) is a method for calculating changes in either binding free energy (of a small molecule or protein binder) or protein stability. Furui & Ohue (1) showed that models from all-atom protein-structure predictors are as accurate as crystal structures for FEP calculations.
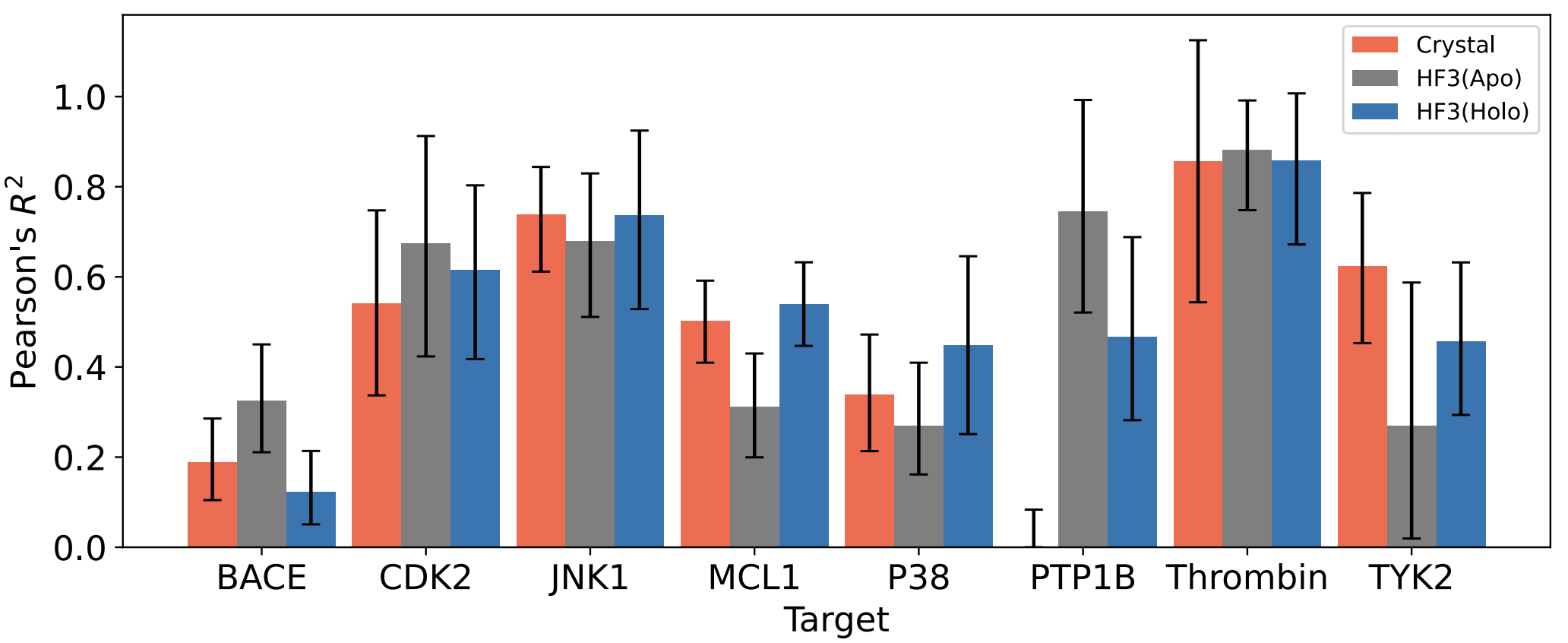 Ref (1)
Ref (1)
Preparing a model for FEP
- Suggested to run at least three short MD simulations prior to FEP to get an appropriate low-energy conformation as a starting point, at least 100 ns but ideally ≥250 ns
- Prior to MD, regions with crystal contacts should be remodeled and refined
Executing FEP
- FEP uses cyclic closure correction, which models changes between mutations (as opposed to just WT→mutant). This allows inaccurate calculations to be identified and corrected.
- The pKa of titratable residues can sometimes change in the bound vs unbound state. This can be accounted for by also performing the simulation using all protonated/deprotonated forms of specific residues (this will also allow cyclic closure correction, see above). Then, the pKa tautomer states can be calculated retrospectively using the following command:
$SCHRODINGER/run -FROM scisol protein_fep_groups.py <jobname>_out.fmp -ph <pH_value> -model_csv model_dG_values.csv. The CSV file is an input containing calculated energy differences between several protonated states of single AAs (model_dG_values.csv). Two output CSV files will be generated with pKa values of each residue in the bound and unbound states. - Using the
-stabilityflag will simultaneously calculate both affinity and stability values.
Summary PPT
All suggestions here were provided courtesy of Dan Cannon and Katalin Phimister at Schrödinger and are relevant to the 2023-3 release of Maestro
1.
Furui K, Ohue M. Benchmarking HelixFold3-Predicted Holo Structures for Relative Free Energy Perturbation Calculations. ACS Omega. 2025;10(11):11411–20. Available from: https://doi.org/10.1021/acsomega.4c11413