Structure prediction refers to the problem of predicting the 3D shape of a protein or nucleotide sequence without any experimental information. Common metrics used for evaluating the quality of predicted structures include LDDT (residue-level, TM-score (whole-structure level), and DockQ (complex level).
Methods
MSA-based
- AlphaFold2: currently viewed as the highest-accuracy method
- RosettaFold
- Diffold: A fine-tuned version of AlphaFold2
PLM-based
- ESMFold: currently the most widely-used method, albeit probably not the most accurate model in this category
- OmegaFold
- xTrimoPGLM
Others
- EquiFold: a method that needs to be fine-tuned on specific families of proteins
- EigenFold: a method that uses diffusion to model the dynamics of proteins, albeit unsuccessfully
For antibodies
See Antibody structure prediction
Notes
Training
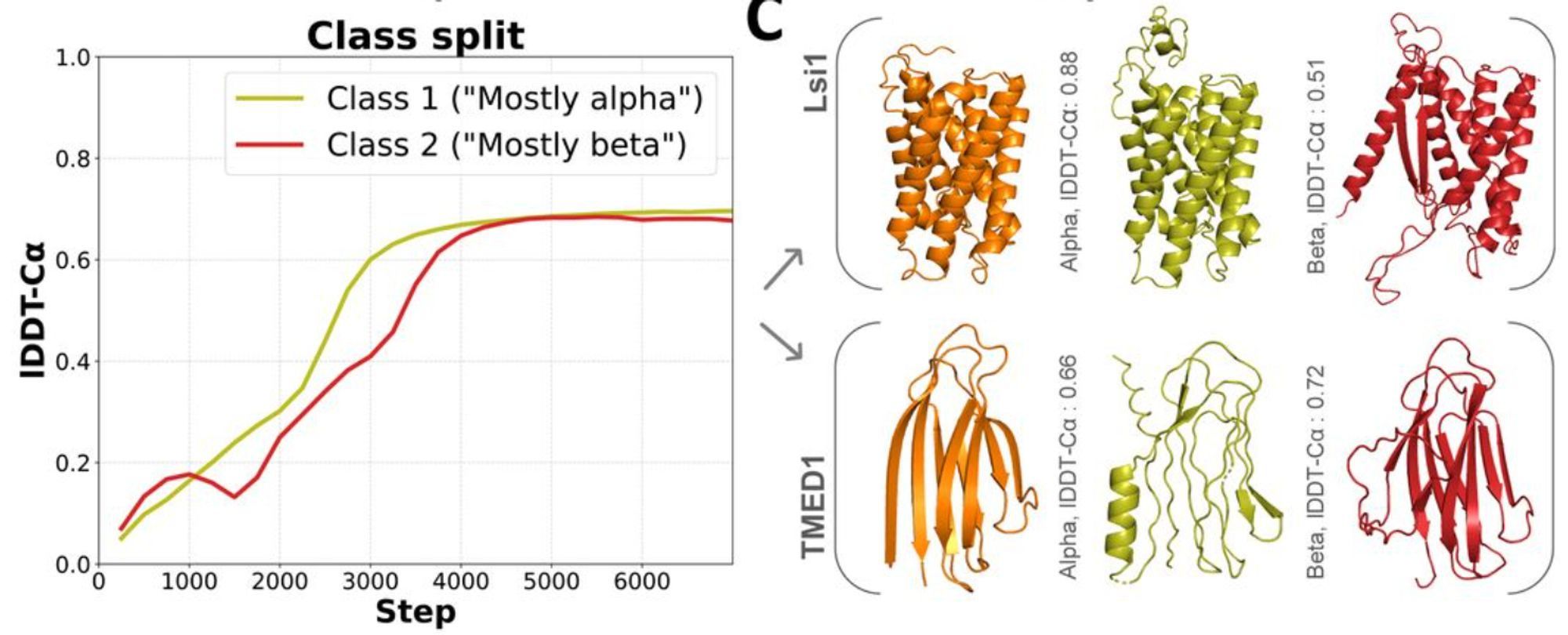 Figure from (1)
Figure from (1)
Sidechain prediction
- Formulating the sidechain prediction problem as a classification problem by binning chi angles, rather than a regression problem, let to improved performance (2).
- Sidechain prediction methods not sensitive to B-factor cutoffs. The outcome of sidechain prediction model PIPPack was not strongly affected by B-factor values of protein structures in the training set (2).
1.
Ahdritz G, Bouatta N, Floristean C, Kadyan S, Xia Q, Gerecke W, et al. OpenFold: retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization. Nature Methods. 2024;21(8):1514–24. Available from: https://doi.org/10.1038/s41592-024-02272-z
2.
Randolph NZ, Kuhlman B. Invariant point message passing for protein side chain packing. Proteins: Structure, Function, and Bioinformatics. 2024;92(10):1220–33. Available from: https://doi.org/10.1002/prot.26705