Summary
High-accuracy computational models might not always be effective for ligand docking. Retrospective screens of AlphaFold2 structures cannot recapitulate ligand binding via Rosetta and GLIDE, even when ligand-binding pockets are correctly modeled ((1); Chris de Graaf’s presentation at the 2022 EMBL AlphaFold workshop). In contrast, prospective screens on two G protein-coupled receptors using AlphaFold models have been successful (2), while (3) found that AlphaFold2 models were comparable in usefulness for virtual screening to apo structures but inferior to holo structures.
Details
This is contrary to work with DiffDock, with 22% of its top ranked ligands within 2Å of the target predicted by ESMFold, which could be due to its ability to “generalize to imperfect structures” (4).
The success of prospective screens by (2) was attributed to the flexibility of the binding pocket of their target:
For the σ2 receptor, 54% of the AF2-derived docking hits were active at 1 µM, while for the crystal structure-derived docking the hit rate was 51%—not statistically different. Meanwhile, for the 5-HT2A receptor, 26% of the molecules from the AF2-derived model bound at 10 µM, while for the cryoEM experimental structure 23% bound.
They found using cryo-EM that the AlphaFold2 model being used for docking was in fact stabilized by a small molecule ligand prospectively obtained by virtual screening. However, the rotamers were mostly or entirely correct, which they found was not always true of GPCRs in the PDB and AFDB.
Figures
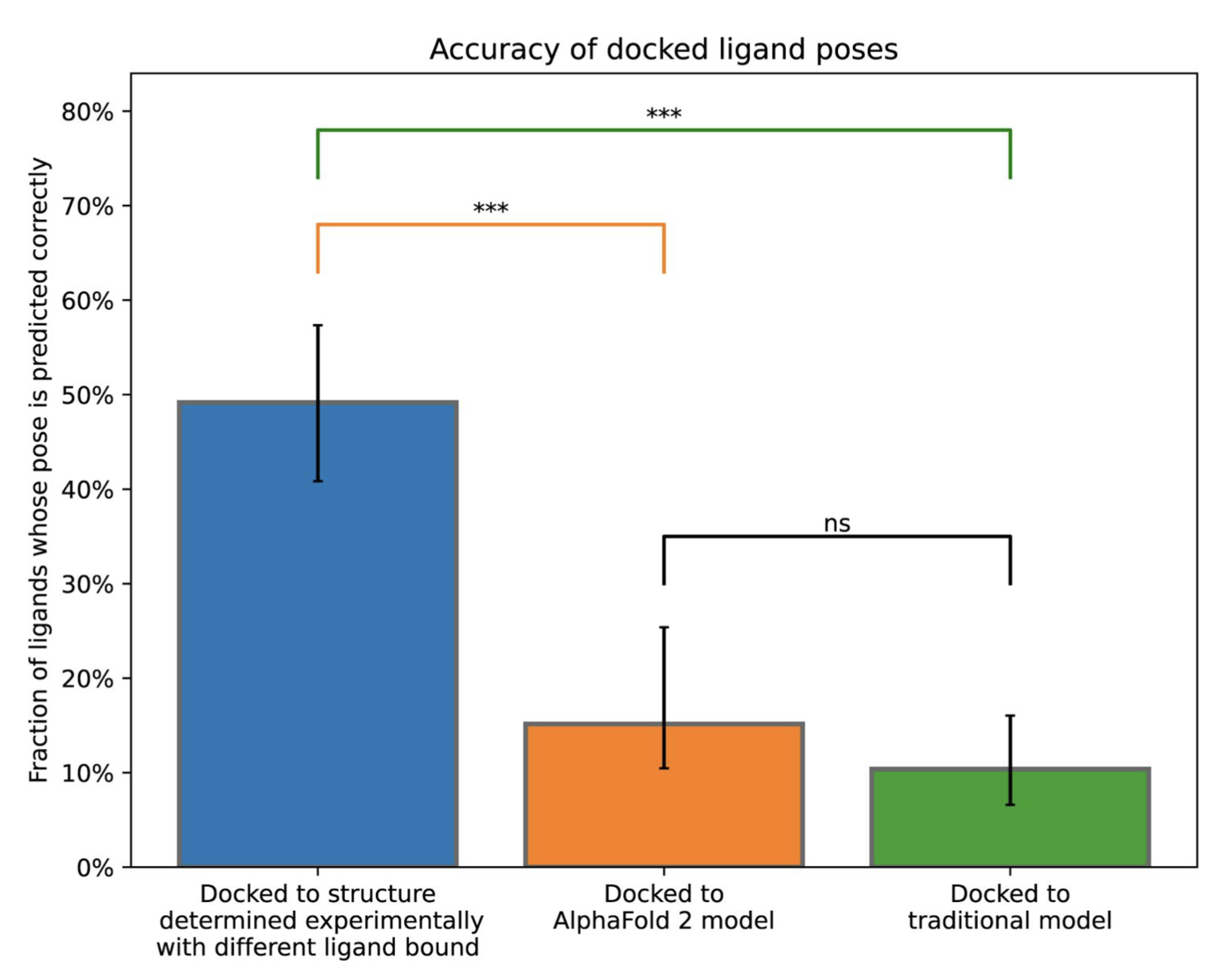 Ref (1)
Ref (1)