Summary
De novo structure prediction with ligands leads to more accurate prediction of structure compared to prediction without ligands (1,2). Predictions of ligand-bound structures using RosettaFold all-atom were on average more accurate than those using default RosettaFold. (1) found that predicted structures without ligands adopted the apo conformation, while those with the ligand adopted the holo conformation. In contrast, AlphaFold2 predictions were somewhere in the middle, and had worse RMSDs/TM-scores for holo but not apo state.
Figures
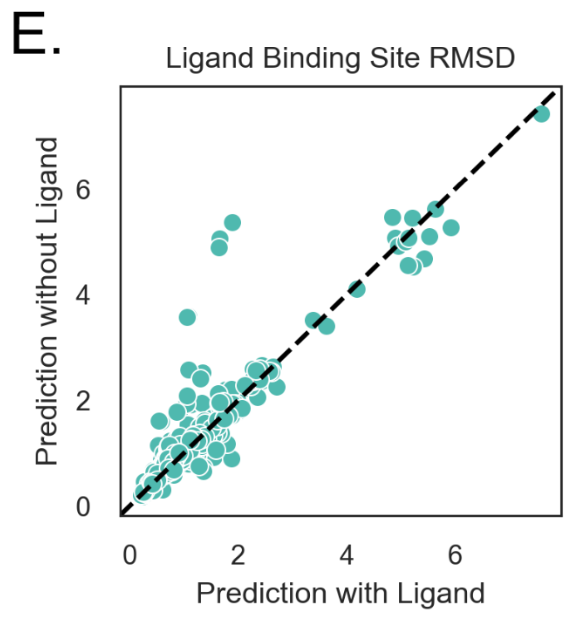
Figure S4E from (2)
See also
- AlphaFold2 predicts the holo form of proteins in most cases
- AlphaFold2 predicts Apo conformations with poorer RMSD than holo conformations
- Including ligands restrains design of catalytically active residues
1.
Qiao Z, Nie W, Vahdat A, Miller TF, Anandkumar A. State-specific protein–ligand complex structure prediction with a multiscale deep generative model. Nature Machine Intelligence. 2024;6(2):195–208. Available from: https://doi.org/10.1038/s42256-024-00792-z
2.
Krishna R, Wang J, Ahern W, Sturmfels P, Venkatesh P, Kalvet I, et al. Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science. 2024;384(6693). Available from: https://doi.org/10.1126/science.adl2528