Summary
Protein structural ensemble prediction methods such as AlphaFlow and ESMFlow, as well as MSA-based tricks such as subsampling and clustering, are unable to capture alternate conformations found in X-ray-crystallography density (1). In contrast, MD simulations can often (but not always) sample both states.
Figures
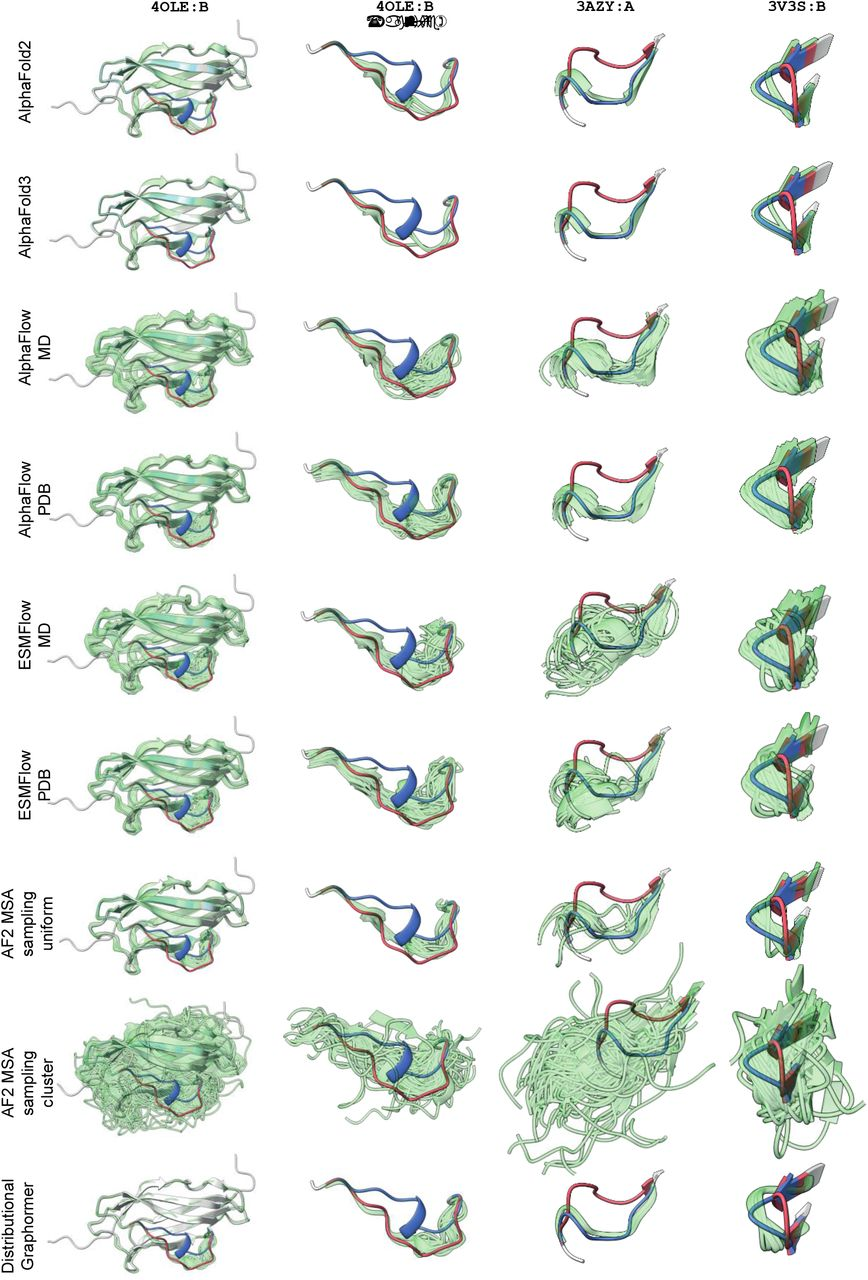 Ref (1)
Ref (1)
See also
1.
Rosenberg AA, Vedula S, Bronstein AM, Marx A. Seeing Double: Molecular dynamics simulations reveal the stability of certain alternate protein conformations in crystal structures. openRxiv; 2024. Available from: https://doi.org/10.1101/2024.08.31.610605