Summary
Introducing noise to structures during training improves self-consistency RMSD of generic inverse folding models but not antibody inverse folding models, while worsening sequence recovery (1–2). It also improves self-consistency RMSD when forward-folding de novo designed proteins (3).
Figures
| Exp/Pred | Layer Decay | OAS Gaussian Noise | Test Masking | FR Avg. | CDR1H | CDR2H | CDR3H | CDR1L | CDR2L | CDR3L |
|---|---|---|---|---|---|---|---|---|---|---|
| Exp | - | - | None | 0.898 | 0.731 | 0.712 | 0.569 | 0.723 | 0.736 | 0.718 |
| Exp | - | ✓ | None | 0.898 | 0.735 | 0.698 | 0.566 | 0.716 | 0.702 | 0.713 |
| Exp | ✓ | - | None | 0.895 | 0.741 | 0.700 | 0.584 | 0.716 | 0.741 | 0.725 |
| Exp | ✓ | ✓ | None | 0.894 | 0.727 | 0.702 | 0.573 | 0.720 | 0.728 | 0.727 |
| Exp | - | - | CDRs | 0.894 | 0.680 | 0.637 | 0.432 | 0.677 | 0.689 | 0.661 |
| Exp | - | ✓ | CDRs | 0.894 | 0.696 | 0.651 | 0.434 | 0.692 | 0.680 | 0.659 |
| Exp | ✓ | - | CDRs | 0.890 | 0.675 | 0.657 | 0.431 | 0.666 | 0.689 | 0.658 |
| Exp | ✓ | ✓ | CDRs | 0.891 | 0.681 | 0.653 | 0.430 | 0.666 | 0.698 | 0.655 |
| Pred | - | - | None | 0.909 | 0.753 | 0.716 | 0.561 | 0.738 | 0.731 | 0.722 |
| Pred | - | ✓ | None | 0.905 | 0.749 | 0.704 | 0.558 | 0.729 | 0.725 | 0.722 |
| Pred | ✓ | - | None | 0.907 | 0.750 | 0.730 | 0.572 | 0.746 | 0.737 | 0.730 |
| Pred | ✓ | ✓ | None | 0.903 | 0.744 | 0.713 | 0.554 | 0.744 | 0.733 | 0.718 |
| Pred | - | - | CDRs | 0.904 | 0.706 | 0.650 | 0.445 | 0.691 | 0.687 | 0.665 |
| Pred | - | ✓ | CDRs | 0.901 | 0.709 | 0.657 | 0.435 | 0.701 | 0.690 | 0.658 |
| Pred | ✓ | - | CDRs | 0.903 | 0.695 | 0.654 | 0.435 | 0.675 | 0.675 | 0.654 |
| Pred | ✓ | ✓ | CDRs | 0.898 | 0.699 | 0.647 | 0.433 | 0.682 | 0.682 | 0.658 |
Table from (5)
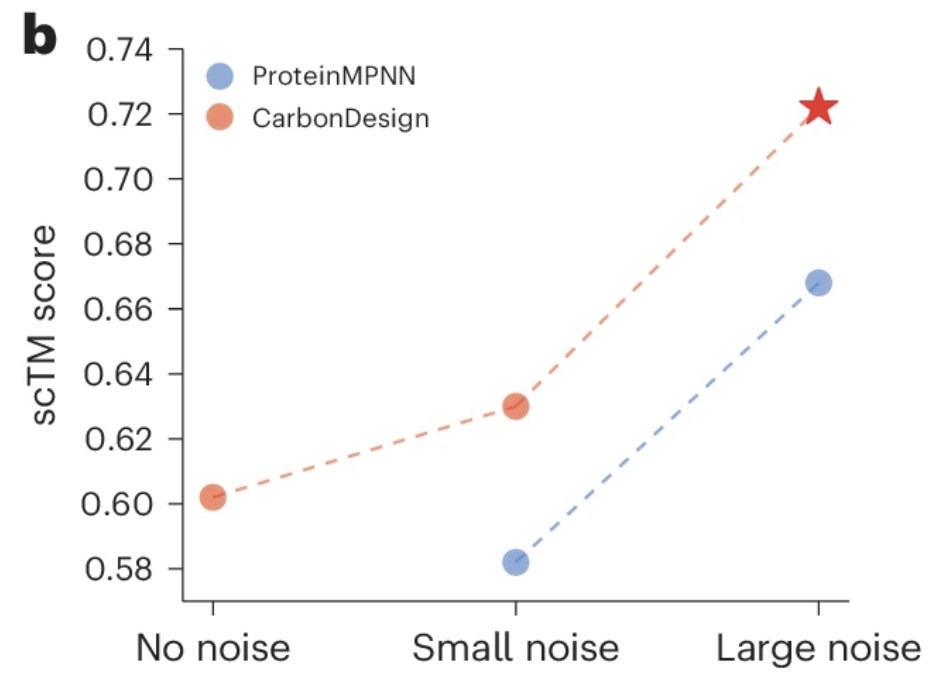
Figure 3b from (3)
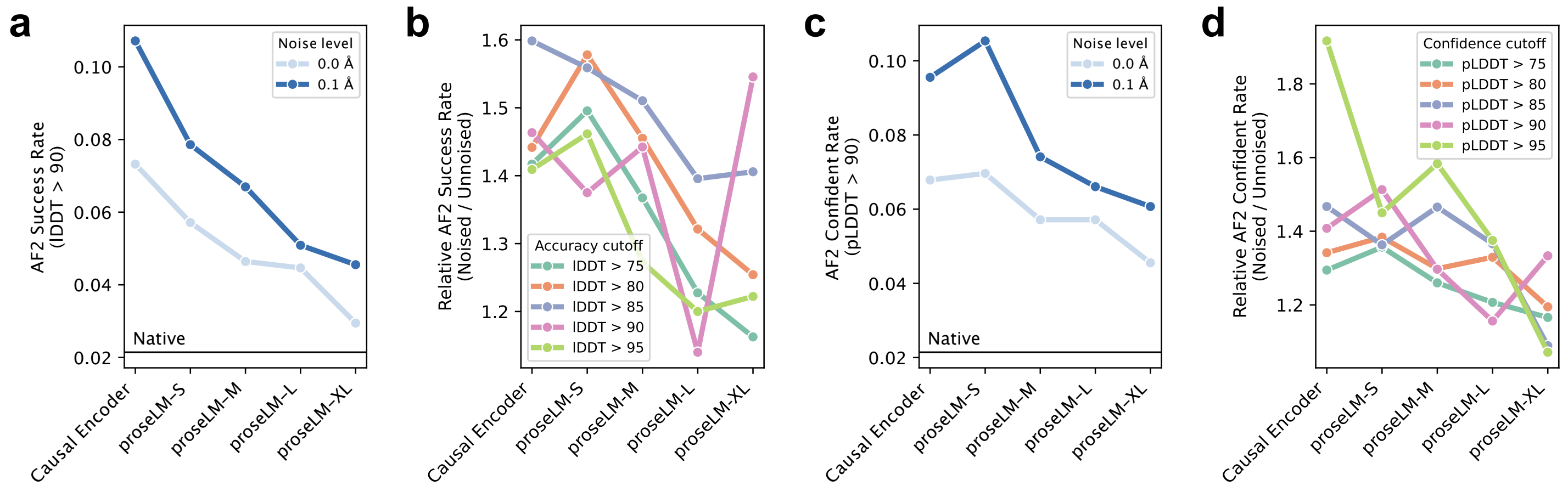
Ref (4)
See also
- ProteinMPNN
- ESM-IF
- Adding noise sometimes leads to greater diversity during inverse folding inference
1.
Dauparas J, Anishchenko I, Bennett N, Bai H, Ragotte RJ, Milles LF, et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science. 2022;378(6615):49–56. Available from: https://doi.org/10.1126/science.add2187
2.
Hsu C, Verkuil R, Liu J, Lin Z, Hie B, Sercu T, et al. Learning inverse folding from millions of predicted structures. openRxiv; 2022. Available from: https://doi.org/10.1101/2022.04.10.487779
3.
Ren M, Yu C, Bu D, Zhang H. Accurate and robust protein sequence design with CarbonDesign. Nature Machine Intelligence. 2024;6(5):536–47. Available from: https://doi.org/10.1038/s42256-024-00838-2
4.
Ruffolo JA, Bhatnagar A, Beazer J, Nayfach S, Russ J, Hill E, et al. Adapting protein language models for structure-conditioned design. openRxiv; 2024. Available from: https://doi.org/10.1101/2024.08.03.606485
5.
Hoie M, Hummer A, Olsen T, Nielsen M, Deane C. AntiFold: Improved antibody structure design using inverse folding. In: GenBio@NeurIPS2023. 2023. Available from: https://openreview.net/forum?id=bxZMKHtlL6