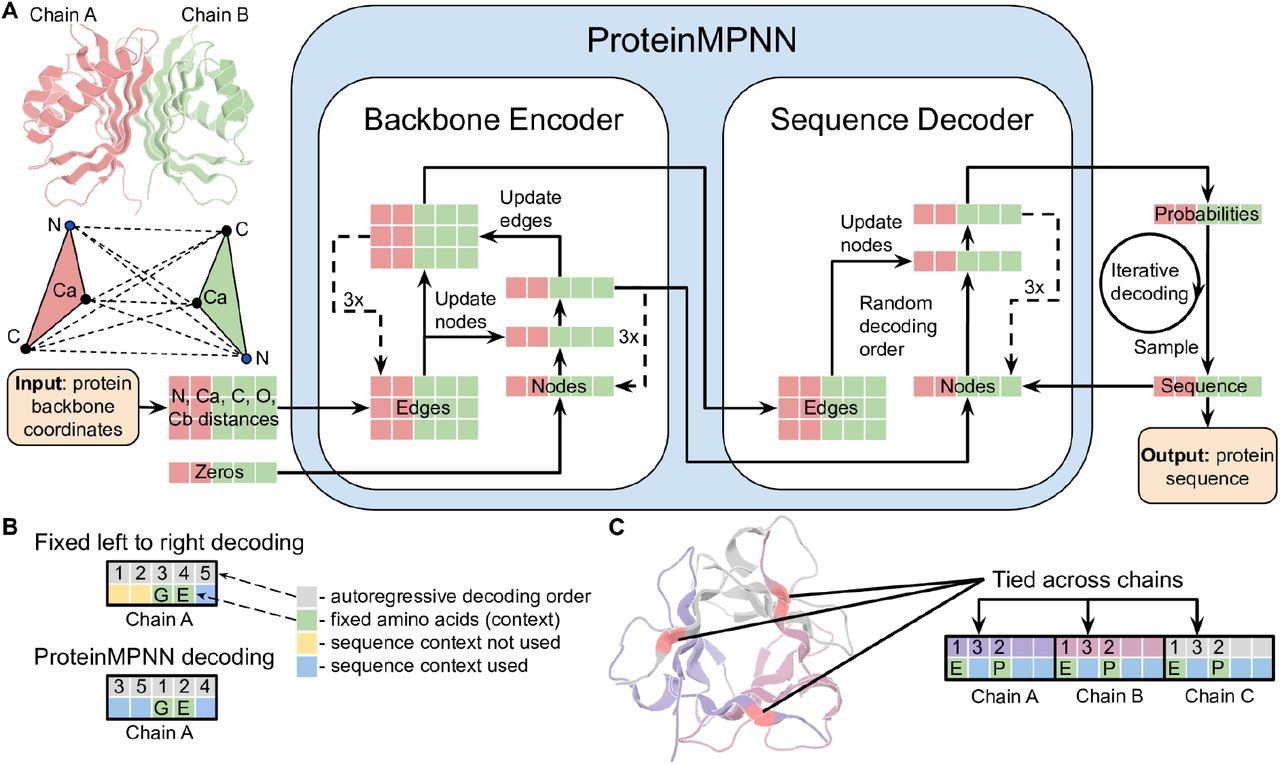
ProteinMPNN is an inverse folding method that uses a message-passing neural network. It has extensive wet-lab validation.
 Ref (1)
Ref (1)
Notes
- ProteinMPNN scores have been shown to correlate poorly with enzyme activity (link).
- Sequence recovery using ProteinMPNN matched the success rate of Rosetta when tasked with designing small protein binders, while being much faster (2).
- Alternating between ProteinMPNN and Rosetta FastRelax improves success when normalized to CPU time, even though it is more expensive per sequence (2).
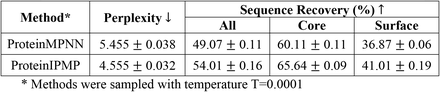
- Sequence recovery can be improved by replacing the message-passing component with Invariant point attention (see ProteinIPMP below; (3)).
- Constrained inverse folding allows design of functional enzymes (4).
- Dauparas et al. (1) found that ProteinMPNN was insensitive to global sequence context. In other words, removing information about sequence adjacency had no effect on sequence recovery. This could be a symptom of Over-squashing.
Variations
- AbMPNN: A retrained version specifically designed for Antibodies (5). Outperforms default ProteinMPNN on several metrics. However it is itself outperformed by AntiFold, trained on ESM-IF (6).
- MiniMPNN: A modified version of ProteinMPNN that has performance; i.e., it predicts the whole sequence in a single pass. Used by ProtPardelle (7).
- ProteinIPMP: A version that uses Invariant point attention, leading to accuracy improvements. Co-released with PIPPack.
- ThermoMPNN: A topped-off version for predicting Stability and thermostability. Trained using a high-quality subset of the Tsuboyama et al. (8) data (9). Was found by Beltran et al. (10) to be the best at variant effect prediction.
- LigandMPNN: A version that can account for non-protein matter (11).
- IgMPNN: A version pretrained on the PDB and fine-tuned on antibody structures (12).
- SoftAlign: A retrained encoder used for structure-based alignment (13).
1.
Dauparas J, Anishchenko I, Bennett N, Bai H, Ragotte RJ, Milles LF, et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science. 2022;378(6615):49–56. Available from: https://doi.org/10.1126/science.add2187
2.
Bennett NR, Coventry B, Goreshnik I, Huang B, Allen A, Vafeados D, et al. Improving de novo protein binder design with deep learning. Nature Communications. 2023;14(1). Available from: https://doi.org/10.1038/s41467-023-38328-5
3.
Randolph NZ, Kuhlman B. Invariant point message passing for protein side chain packing. Proteins: Structure, Function, and Bioinformatics. 2024;92(10):1220–33. Available from: https://doi.org/10.1002/prot.26705
4.
Sumida KH, Núñez-Franco R, Kalvet I, Pellock SJ, Wicky BIM, Milles LF, et al. Improving Protein Expression, Stability, and Function with ProteinMPNN. Journal of the American Chemical Society. 2024;146(3):2054–61. Available from: https://doi.org/10.1021/jacs.3c10941
5.
Dreyer FA, Cutting D, Schneider C, Kenlay H, Deane CM. Inverse folding for antibody sequence design using deep learning. 2023; Available from: https://arxiv.org/abs/2310.19513
6.
Hoie M, Hummer A, Olsen T, Nielsen M, Deane C. AntiFold: Improved antibody structure design using inverse folding. In: GenBio@NeurIPS2023. 2023. Available from: https://openreview.net/forum?id=bxZMKHtlL6
7.
Chu AE, Kim J, Cheng L, El Nesr G, Xu M, Shuai RW, et al. An all-atom protein generative model. Proceedings of the National Academy of Sciences. 2024;121(27). Available from: https://doi.org/10.1073/pnas.2311500121
8.
Tsuboyama K, Dauparas J, Chen J, Laine E, Mohseni Behbahani Y, Weinstein JJ, et al. Mega-scale experimental analysis of protein folding stability in biology and design. Nature. 2023;620(7973):434–44. Available from: https://doi.org/10.1038/s41586-023-06328-6
9.
Dieckhaus H, Brocidiacono M, Randolph NZ, Kuhlman B. Transfer learning to leverage larger datasets for improved prediction of protein stability changes. Proceedings of the National Academy of Sciences. 2024;121(6). Available from: https://doi.org/10.1073/pnas.2314853121
10.
Beltran A, Jiang X, Shen Y, Lehner B. Site saturation mutagenesis of 500 human protein domains reveals the contribution of protein destabilization to genetic disease. openRxiv; 2024. Available from: https://doi.org/10.1101/2024.04.26.591310
11.
Dauparas J, Lee GR, Pecoraro R, An L, Anishchenko I, Glasscock C, et al. Atomic context-conditioned protein sequence design using LigandMPNN. Nature Methods. 2025;22(4):717–23. Available from: https://doi.org/10.1038/s41592-025-02626-1
12.
Shanehsazzadeh A, Alverio J, Kasun G, Levine S, Calman I, Khan JA, et al. IgDesign: In vitro validated antibody design against multiple therapeutic antigens using inverse folding. openRxiv; 2023. Available from: https://doi.org/10.1101/2023.12.08.570889
13.
Trinquier J, Petti S, Park S, Herath K, van Kempen M, Feng S, et al. SoftAlign: End-to-end protein structures alignment. openRxiv; 2025. Available from: https://doi.org/10.1101/2025.05.09.653096