Summary
Training machine learning models for either inverse folding or protein backbone design via diffusion exclusively on predicted models worsens performance (1,2). This was observed when training ESM-IF and GVP as well as when training using the (Evoformer) or the hybrid sequence-structure method MIF-ST, but not (SaProt) (which uses tokens from the (Foldseek) alphabet). The latter study also looked at downstream performance and saw worse results. This was shown to be because predicted models are “too perfect” at a local level (3).
Details
(4) found that training their diffusion model on both predicted models and experimental structures worsened designability and novelty relative to a model trained on experimental structures only.
(5) trained a version of the backbone diffusion model GENIE on (AlphaFold2) models from SwissProt and found that although designability increased was greater than a model trained on the PDB, diversity was lower.
Figures
| Exp only | AF2+Exp | AF2 only | |
|---|---|---|---|
| GVP-GNN | 5.43 | 6.06 | 6.52 |
| GVP-GNN-Large | 6.17 | 4.08 | 11.51 |
| GVP-Transformer | 6.44 | 4.01 | 10.95 |
Table from (1)
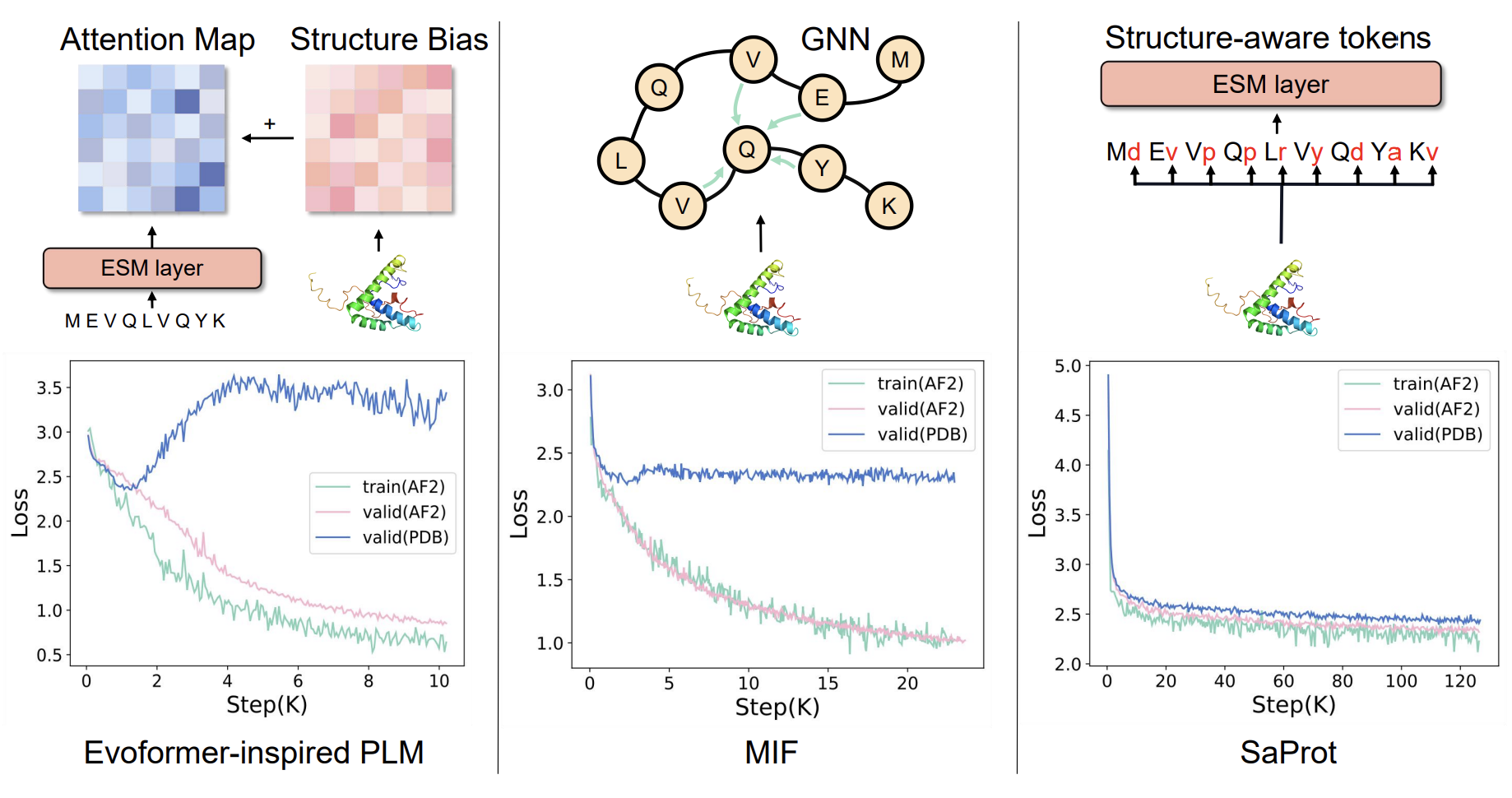 Ref (2)
Ref (2)
![[bafkreiawfxhyqc4grpfhhgjsyezzahtsrehsxzughw6vmpsuw2tqsazz64@jpeg.jpg]]
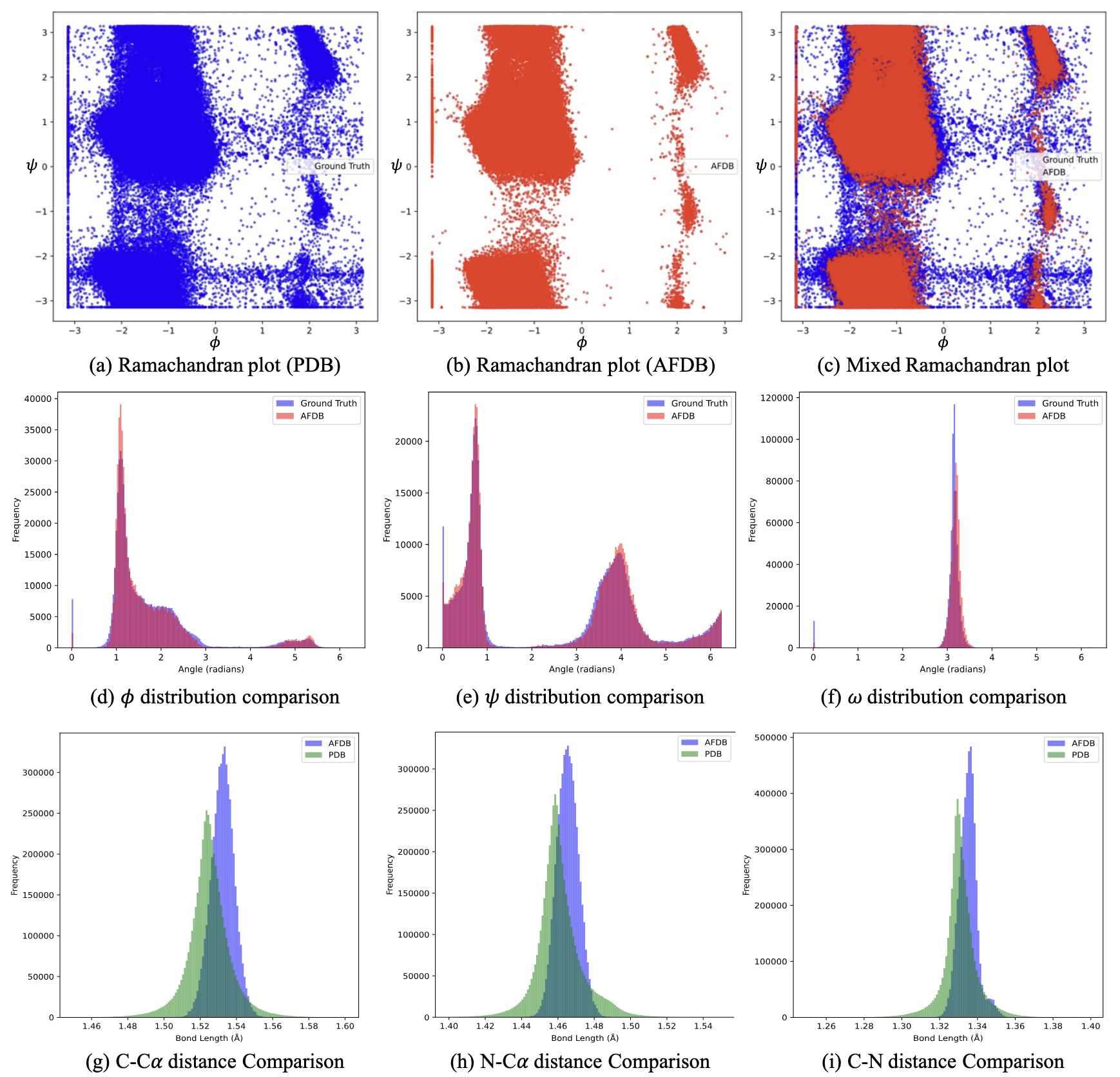 Figures from (3)
Figures from (3)
{kind=link}
See also
- Inverse folding models trained on all proteins outperform those trained on Abs for CDR prediction
- Adding noise while training non-Ab inverse folding models improves self-consistency while worsening sequence recovery
- Focused protein sequence libraries are poor training sets
- Computational models are less designable than experimental structures