Inverse folding describes the problem of designing a sequence for a structure. Typically these are limited to the twenty canonical amino acids.
Methods
See Hybrid sequence-structure models for a list of methods that incorporate PLMs
- ProteinMPNN and its derivatives
- ESM-IF
- GearNet
- Frame2seq
- GVP-GNN
Notes
Training
- Training inverse folding models with backbone dihedral angles as features usually improved sequence recovery (1).
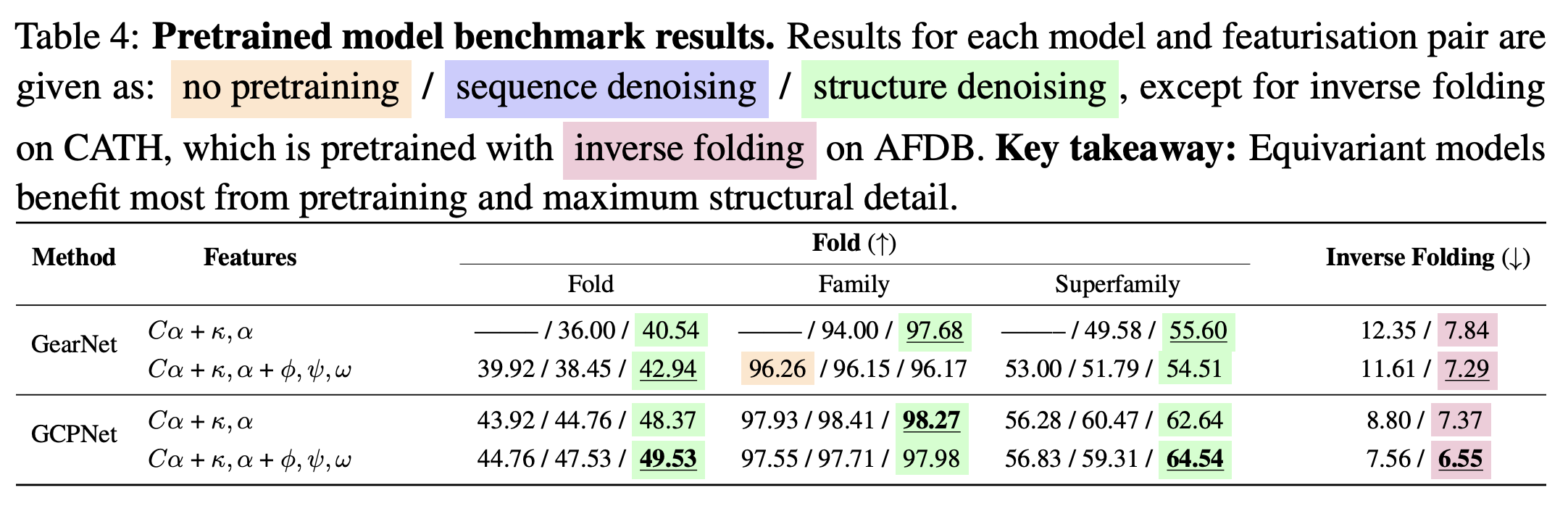 Figure from (1)
Figure from (1)
Execution
- Forward-folding is a stronger predictor of inverse folding success than sequence recovery ((2), citing Watson et al. (3,4)).
Datasets
- PDBench is a dataset of 595 protein structures with diverse, evenly divided topologies for benchmarking of Inverse folding methods (5).
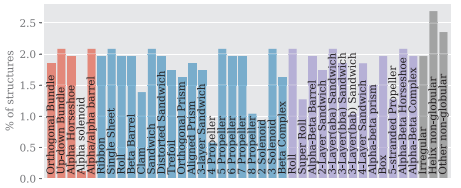 Figure 2 from Castorina et al. (5)
Figure 2 from Castorina et al. (5)
1.
Jamasb AR, Morehead A, Joshi CK, Zhang Z, Didi K, Mathis SV, et al. Evaluating Representation Learning on the Protein Structure Universe. In: ICLR 2024. 2024. Available from: https://openreview.net/forum?id=sTYuRVrdK3
2.
Yang JJ, Yim J, Barzilay R, Jaakkola T. Fast non-autoregressive inverse folding with discrete diffusion. 2023; Available from: https://arxiv.org/abs/2312.02447
3.
Watson JL, Juergens D, Bennett NR, Trippe BL, Yim J, Eisenach HE, et al. De novo design of protein structure and function with RFdiffusion. Nature. 2023;620(7976):1089–100. Available from: https://doi.org/10.1038/s41586-023-06415-8
4.
Dauparas J, Anishchenko I, Bennett N, Bai H, Ragotte RJ, Milles LF, et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science. 2022;378(6615):49–56. Available from: https://doi.org/10.1126/science.add2187
5.
Castorina LV, Petrenas R, Subr K, Wood CW. PDBench: evaluating computational methods for protein-sequence design. Bioinformatics. 2023;39(1). Available from: https://doi.org/10.1093/bioinformatics/btad027