Summary
Alternate conformations can be sampled with MSA-based methods AlphaFold2 and Boltz by using either custom templates (1) or custom sequences databases (2) or MSAs (3,1). For the last case, MSAs are modified either by clustering using HDBSCAN or randomly subsampling, respectively. However, these ensembles do not correspond to the energetics of those proteins. MSA-based tricks were recently shown to work with Boltz (4). These hacks circumvent the broader tendency of structure prediction methods to undersample conformations they find to be high-confidence.
Details
Alternate conformations sampled via
- Subsampled MSAs: Fewer sequences are passed in the MSA (1). Required for addition of other restraints (e.g., via AlphaLink, (5)). Also works for CDRs of nanobodies (6).
- Masked MSA columns: Entire columns of residues are mutated to either alanine or masked (7,8)
- Clustered MSAs: (3)
- Template curation: Can more reliably influence which conformation is sampled, but requires an experimental structure (though Faezov and (9) used models for some of their kinases).
- Dropout: Shown by (8) to not be very effective, unlike for docking (link)
(10) Vani et al summarized the challenge of MSA subsampling as follows:
“The structures obtained, including those that are metastable, are not in any physically reasonable probability distribution. Nor is there an obvious way to directly obtain a distribution or free energy surface from them that could account for both enthalpy and entropy.”
Figures
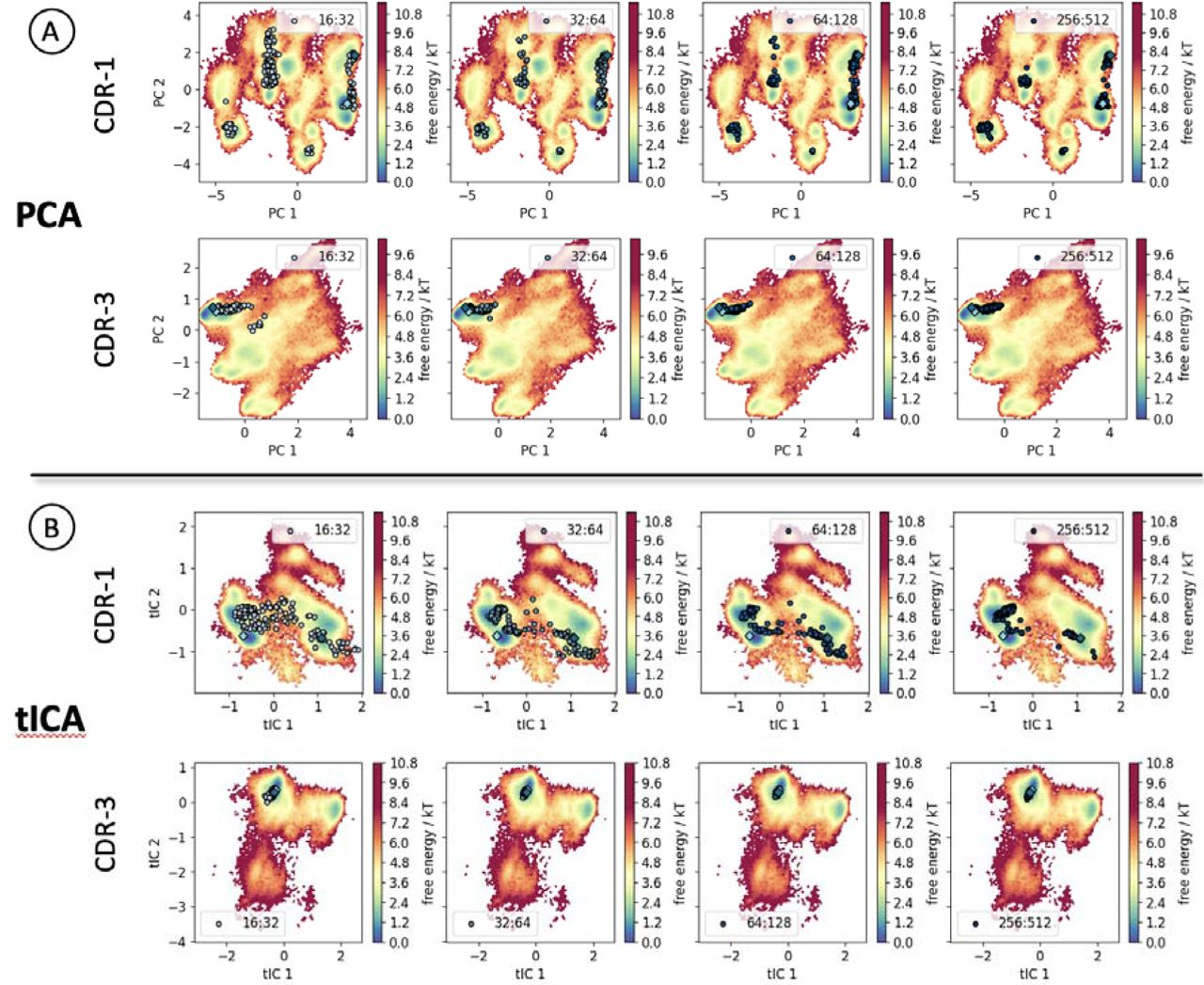
Ref (6)
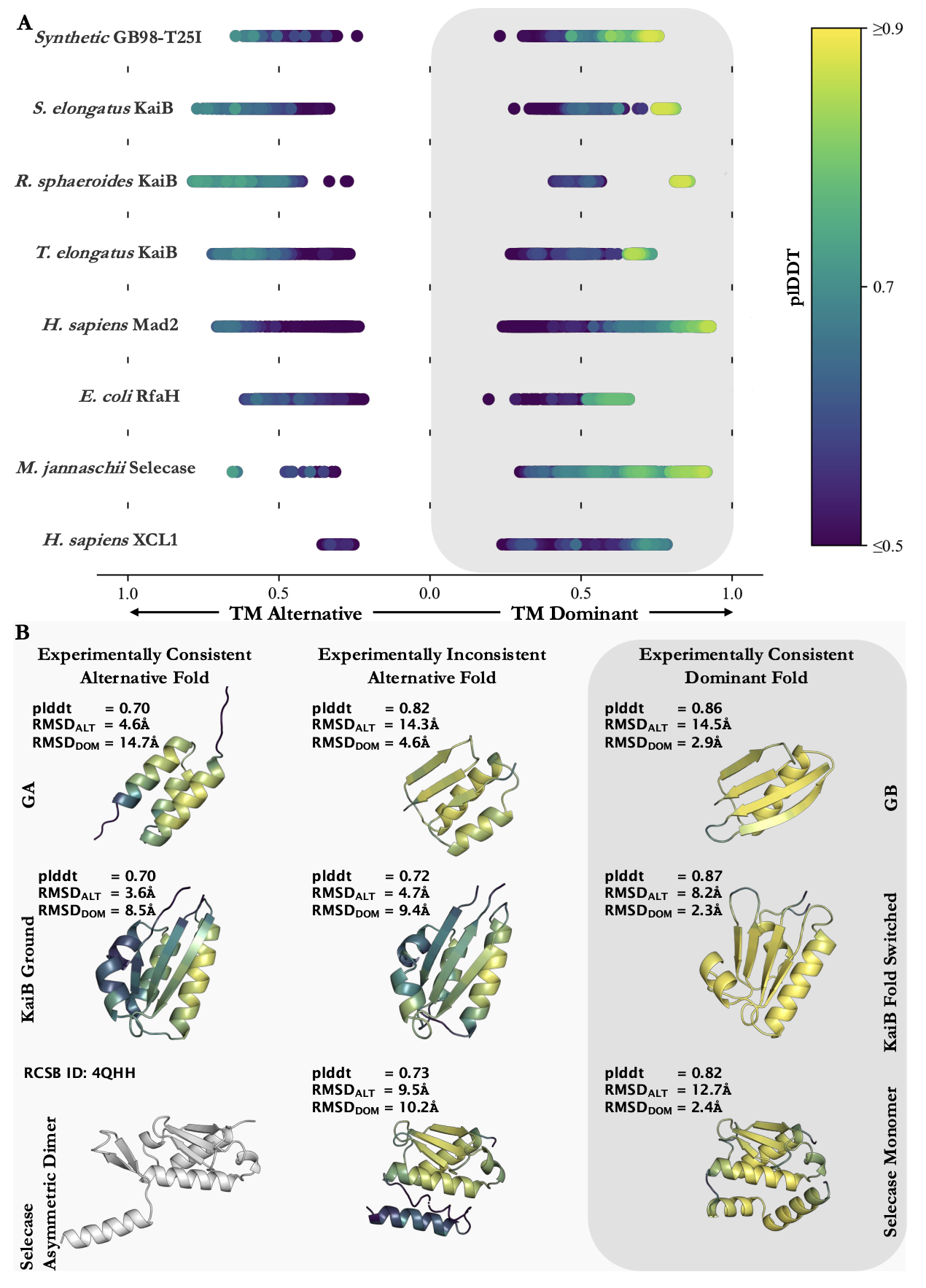
Ref (11)