Summary
Sequence perplexity is a metric used by protein language models and inverse folding to quantify sequence recovery. Self-consistency perplexity is a derived metric where the perplexity is calculated using a forward-folded model rather than the original model/structure.
Details
The perplexity values of null models was calculated below by (1):
 Table from (1)
Table from (1)
Hie et al. found that sequence length does not consistently correlate with perplexity values within protein families. (2)
Meier et al. introduced ways of calculating probabilities or “energies” of sequences using masked LMs (e.g., how favored or disfavored they are given evolution). The authors of ESM-1v propose four approaches for this: (3)
- Masked marginal probability: Masks are introduced at all mutation residues and probabilities relative to the wildtype residue are calculated:
- Mutant marginal probability: Requires a single forward pass for each mutation: . The entire sequence is kept fixed except for one mutation and the preference for the mutation of interest, normalized to the WT residue, is calculated. This was used for structure-based fitness prediction in (4) with Spearman values ranging from 0.39–0.71.
- Wildtype marginal probability: The same as mutant marginal probability, except the background is the wildtype sequence:
- Pseudo-likelihood: (5) found that ESM2 pseudo-log likelihood values scaled linearly with the length of the sequence. They introduced a length-invariant correction (): ,
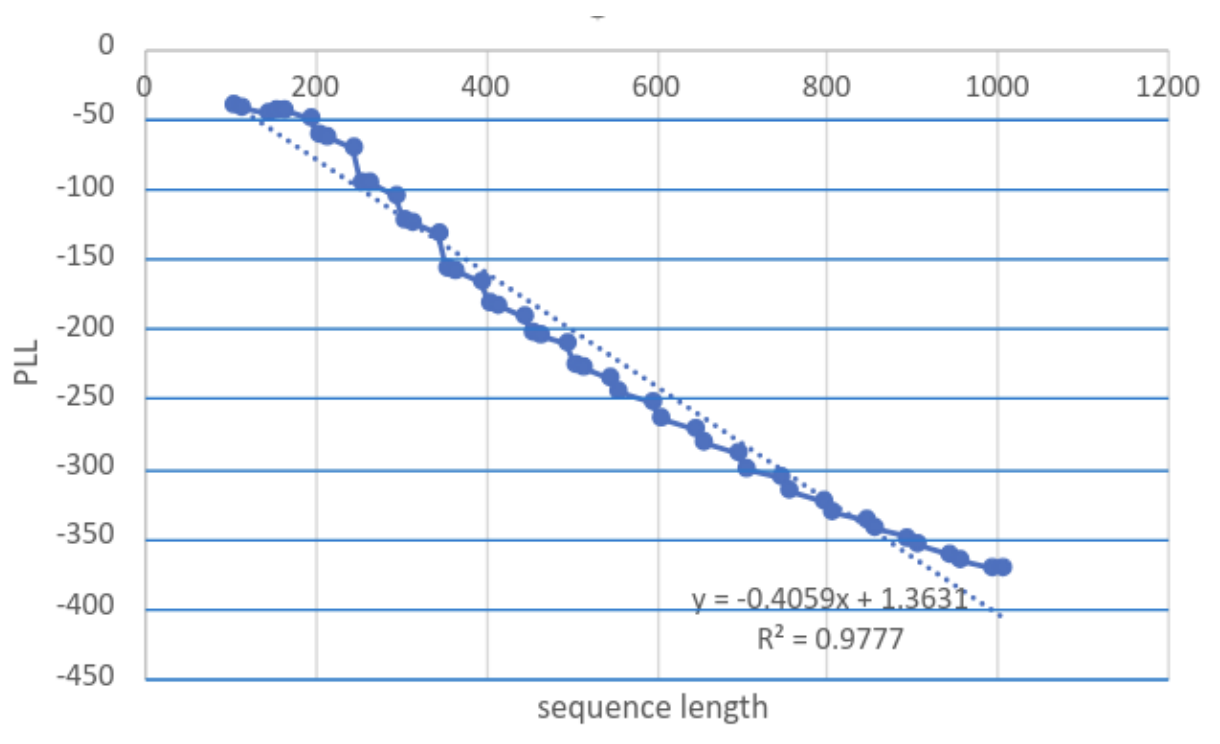 Ref (5)
Ref (5)
The Spearman correlations against ProteinGym were: masked marginal 0.582, mutant marginal 0.578, wildtype marginal 0.572, pseudolikelihood 0.552.
Gordon et al. propose a way to approximate sequence log-likelihood in a single pass using masked LMs: (6)

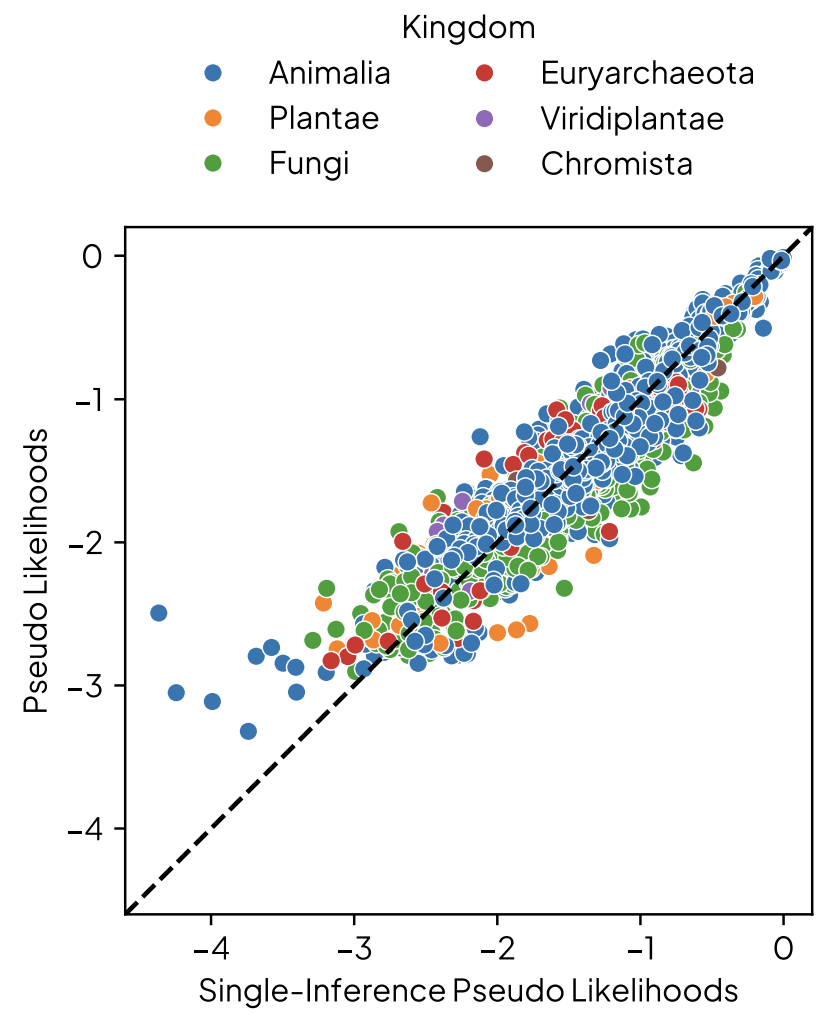 Figures from (6)
Figures from (6)
See also
- Self-consistency perplexity is correlated with pLDDT
- Sequence perplexity and TM-score are negatively correlated when predicting structure using protein language models
- Spearman correlations of protein property prediction methods do not correlate perfectly with absolute error
- Fréchet distance