Tag Index
Found 109 total tags.
affinity-maturation
Affinity maturation is a process by which antibodies are iteratively improved in vivo. Described as “hyper-darwinian”.
29 items with this tag. Showing first 10 tags.
alignment
Alignment collects notes on comparing proteins by either sequence-derived representations or structural information. The main split here is between sequence-based alignment approaches, including PLM embedding methods and homology retrieval, and structure-based alignment approaches that operate on coordinates, structural descriptors, or structure-derived embeddings.
18 items with this tag. Showing first 10 tags.
alignment/sequence-based
14 items with this tag. Showing first 10 tags.
alignment/structure-based
6 items with this tag.
alphafold2
(OpenFold redirects here)
AlphaFold2 is a protein structure prediction method from 2020, and is the first to make extensive use of the Transformer architecture, consisting of 97 million parameters. AlphaMissense is a derivative of this method.
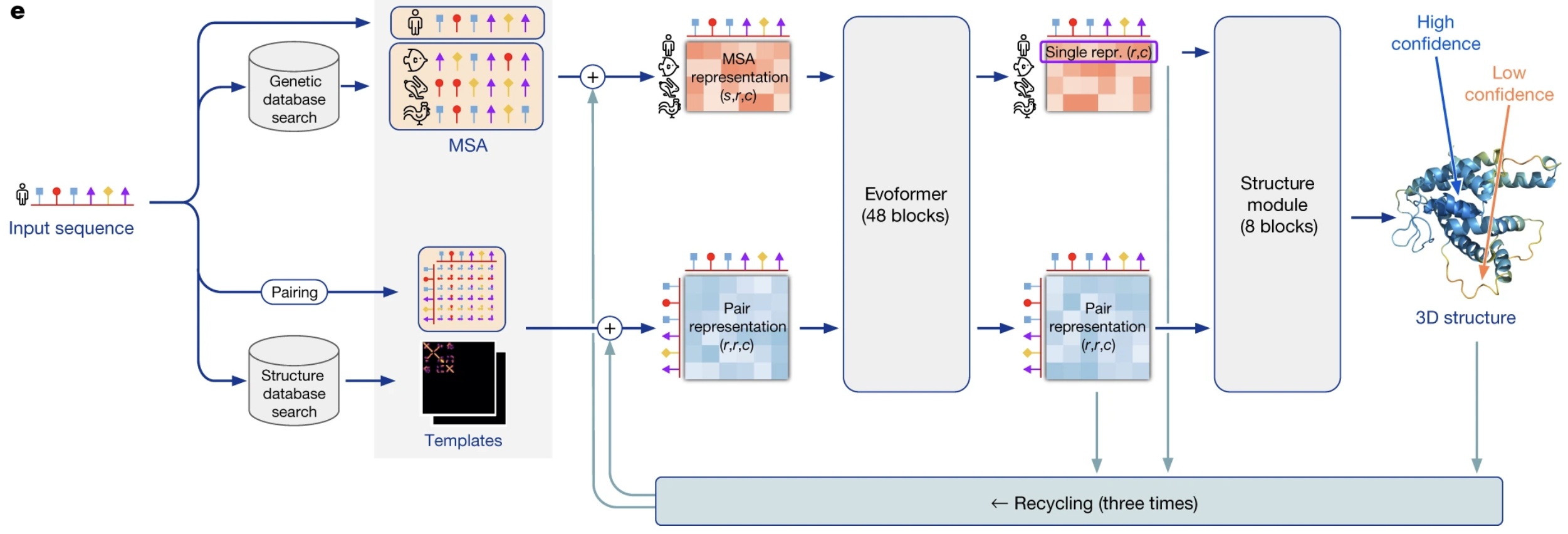 Architecture of AlphaFold2 from Jumper et al. (1)
Architecture of AlphaFold2 from Jumper et al. (1)
Architectural and ML contributions
- Evoformer
- Invariant point attention
- Frame aligned point error
- Self-distillation for protein structures
- Predicted aligned error
- Triangular update
26 items with this tag. Showing first 10 tags.
alphafold3
AlphaFold3 is a diffusion-based all-atom structure prediction method that is widely seen as state-of-the-art.
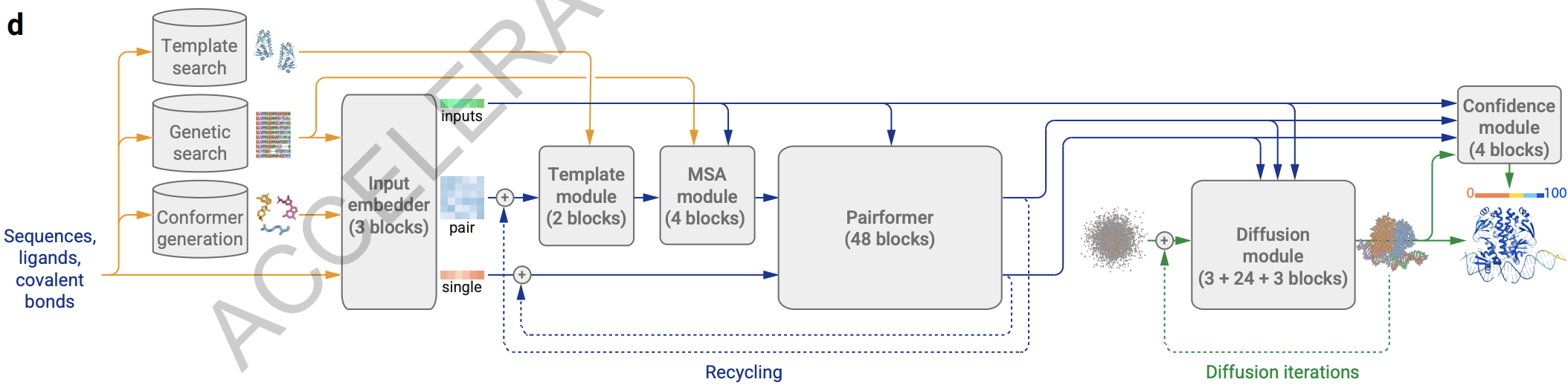 Architecture of AlphaFold3 from Abramson et al. (1)
Architecture of AlphaFold3 from Abramson et al. (1)
Architectural and ML contributions
- PairFormer
- PDE: predicted distance error (replacing frame aligned point error)
- Non-equivariant per-atom prediction, which leads to occasional errors when predicting chirality
- Cross-distillation from AlphaFold2-Multimer v2.3 to avoid hallucination of low-pLDDT regions
24 items with this tag. Showing first 10 tags.
ancestral-sequence-reconstruction
Ancestral sequence reconstruction refers to the process of inferring ancestral sequences using extant sequences.
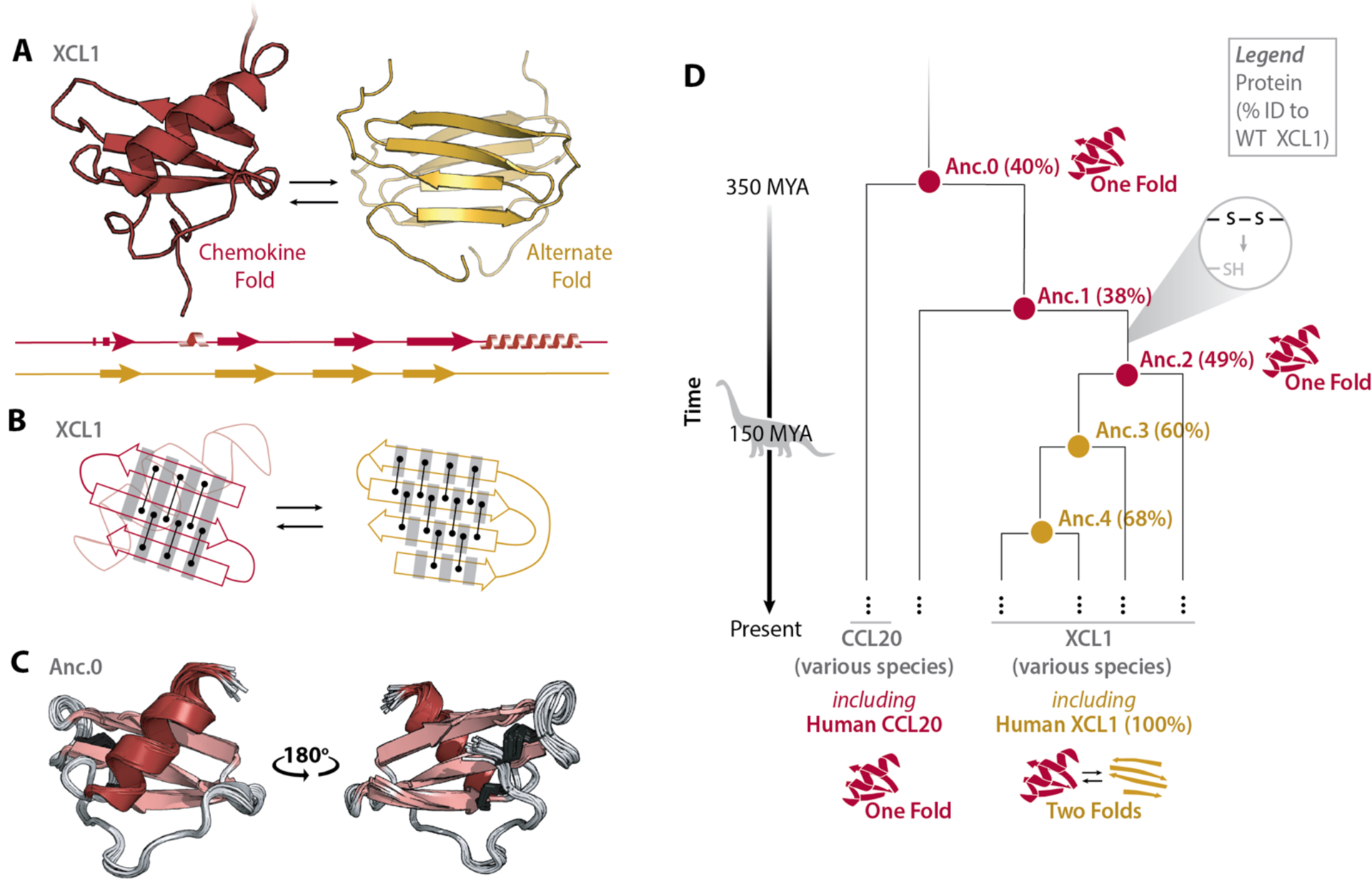 Figure from (1)
Figure from (1)
6 items with this tag.
antibodies
Antibodies are proteins with two heavy chains and two light chains produced by B cells, central to the adaptive immune system. Their structure consists of a variable region (containing CDRs and a framework region) and three constant regions (CH1, CH2, CH3). The variable region and CH1 form the Fab, while the remainder forms the Fc region. B cell receptors are antibodies with an additional CH4 domain.
Types of antibodies
IgA is found in mucous membranes, cannot activate the complement system, and makes up roughly two-thirds of antibodies in healthy adults. It is notable for being double-sided.
IgE has only one binding site, mainly protects against large organisms like parasites, and is responsible for allergic reactions.
IgG is the standard antibody class used in therapeutic design. Cannot activate the complement system and can pass through the placenta.
- IgG1 makes up 67% of all antibodies in the human body, is capable of antibody-dependent cellular phagocytosis, and has the longest hinge region. IgG1 immune repertoires consist mostly of just a few dozen dominant clones and are unique to each individual, remaining largely stable over time.
- IgG2 — mice have IgG2a and IgG2b instead; some strains have IgG2c instead of IgG2a.
- IgG3 makes up ~7% of IgGs and has a notably shorter half-life due to poor binding to FcRn, attributed to an H435R mutation.
- IgG4 is the rarest IgG and undergoes Fab-arm exchange, dissociating into half-bodies and forming novel combinations via R409 in the hinge. Therapeutic IgG4 antibodies use the S228P substitution to stabilize the hinge and prevent this.
IgM is far less subject to affinity maturation than IgG, responds to lipids and polysaccharides, and activates the complement system.
Datasets
SAbDab is a database of all antibody and nanobody structures in the PDB. The full list can be downloaded with:
curl -s https://opig.stats.ox.ac.uk/webapps/sabdab-sabpred/sabdab/summary/all/28 items with this tag. Showing first 10 tags.
antibodies/engineering-and-design
8 items with this tag.
antibodies/function
1 item with this tag.
antibodies/nanobodies
17 items with this tag. Showing first 10 tags.
antibody-antigen-interactions
48 items with this tag. Showing first 10 tags.
antibody-antigen-interactions/binding-affinity
15 items with this tag. Showing first 10 tags.
antibody-antigen-interactions/complex-prediction
18 items with this tag. Showing first 10 tags.
antibody-antigen-interactions/misc
15 items with this tag. Showing first 10 tags.
antibody-developability
Antibody developability refers to a series of properties linked to the development of antibodies as therapeutics. These arise because natural antibodies do not possess them to begin with, or because they arise during optimization as a consequence of affinity maturation via techniques such as Yeast display.
General observations
- Raybould et al. (1) outlined five criteria (in a parallel to Lepinski’s rule of five):
- Length of CDRs, specifically CDRH3
- Hydrophobic patches
- Negative patches
- Positive patches
- Viscosity
Thermostability
See Stability and thermostability
Immunogenicity
- Glycine residues doesn’t cause immunogenicity as much as other residues (2). This makes them used for including during design of cyclic peptides or peptide therapeutics.
- Antibody humanization can lead to immunogenic reactions.
32 items with this tag. Showing first 10 tags.
antibody-developability/expression
4 items with this tag.
antibody-developability/general
10 items with this tag.
antibody-developability/hydrophobicity
5 items with this tag.
antibody-developability/polyspecificity
6 items with this tag.
antibody-drug-conjugates
1 item with this tag.
antibody-structure-prediction
The antibody structure prediction problem is a subclass of the protein structure prediction problem that is mostly focused on predicting the CDRs, specifically the CDRH3 loop that mediates antigen binding. It also sometimes includes the antibody-antigen docking problem.
Models
- AbFold: a method that concatenates information from IgFold and AlphaFold2 Multimer when modeling antibodies, which usually predict different conformations, leading to improved prediction quality (1)
- IgFold: a method that uses embeddings from the antibody-specific PLM AntiBERTy
- ImmuneBuilder: Consists of ABodyBuilder2 (for Antibodies), NanoBodyBuilder2 (for Nanobodies), and TCRBuilder2 (for T-cell receptors).
27 items with this tag. Showing first 10 tags.
antibody-structure-prediction/cdr
19 items with this tag. Showing first 10 tags.
antibody-structure-prediction/complex-prediction
5 items with this tag.
b-cells
B cells are a type of lymphocyte that synthesizes and secretes antibodies as well as B-cell receptors. Each cell synthesizes a single antibody sequence throughout its life cycle (a phenomenon termed allelic exclusion). Can be categorized as either naive B cells or memory B cells. Activation by antigen presentation causes them to proliferate into effector cells that secrete antibodies.
1 item with this tag.
blosum62
The BLOSUM62 matrix quantifies the similarity of amino acids to one another when computing evolutionary distance sequences (1). Interestingly, it has math errors in its computation that make it a more effective matrix for substitution calculation (2). A custom version for TCRs has also been presented (3).
1 item with this tag.
cdrh3
13 items with this tag. Showing first 10 tags.
citation-fix
25 items with this tag. Showing first 10 tags.
citation-needed
1 item with this tag.
confidence-metrics
9 items with this tag.
conformational-dynamics
64 items with this tag. Showing first 10 tags.
conformational-dynamics/allostery
7 items with this tag.
conformational-dynamics/evolution
10 items with this tag.
conformational-dynamics/experimental-ensembles
5 items with this tag.
conformational-dynamics/kinetics
6 items with this tag.
conformational-dynamics/modeling
23 items with this tag. Showing first 10 tags.
conformational-dynamics/molecular-dynamics
12 items with this tag. Showing first 10 tags.
contrastive-learning
Contrastive learning is a supervised method for driving the latent representations of data points towards or away from each other by adding custom losses.
Implementations
- The triplet margin loss used by Yu et al. (1): : Enzyme embedding : Positive case : Negative case, selected to have EC numbers close in Euclidean space to the positive case : Margin; set to 1
- The supercon hard loss used by Yu et al. (1): : temperature, set to 0.1 in the paper
- Noise contrastive estimation (variant 1): : Input text : Another example (either positive or negative) : Scoring function, usually cosine similarity, dot product, or logit “produced by input-sample matcher sub-network” (from Rethmeier and Augenstein 2021) : Scaling function, usually sigmoid
- Noise contrastive estimation (variant 2, ranks a single positive pair over negative pairs): Variant 2 ranks a single positive pair over negative pairs
5 items with this tag.
diffusion-guidance
Diffusion guidance refers to inference-time methods that steer a diffusion process toward desired properties, constraints, or observations. It is a general concept that applies to both protein design, structure prediction, and sequence-based protein design, of which all-atom diffusion is just one application.
Details
Xie et al (1) outline three broad types of guidance used by diffusion models:
- Score guidance, in which gradients from classifiers, constraints, or rewards are used to nudge the diffusion path.
- Path-integral reweighting, such as Feynman-Kac potentials, which use importance weights to update trajectories.
- Invariant correctors, such as Metropolis-adjusted Langevin methods, which mix within a biased marginal without changing the trajectory weights.
However, other search algorithms such as Beam search and Monte Carlo Tree Search have been used in conjunction with diffusion models (2).
12 items with this tag. Showing first 10 tags.
diffusion-guidance/protein-design
5 items with this tag.
diffusion-guidance/structure-prediction
8 items with this tag.
diffusion-models
Diffusion models are generative models whose outputs are generated by iteratively denoising Gaussian noise. During inference, the reverse process is executed, whereas during training, the forward process is carried out (converting data to noise). This process can be guided at inference time to respect specific geometric constraints.
Details
Diffusion models try to recover samples drawn from from Gaussian noise by iteratively denoising across time steps. The forward process is defined as
: Standard Weiner process (by which noise is added)
Typically these drift and diffusion functions are used:
The reverse process:
The scoring function is approximated by a neural network, .
Diffusion models can also be zero-shot classifiers (1).
Diffusion models have been combined with Replica-exchange molecular dynamics (2).
Types of sequence-based diffusion
For protein sequences, which are fundamentally discrete, Yang et al (3) describe three approaches to generation using diffusion models:
- Diffusion in pre-trained latent space (e.g., continuous diffusion)
- Diffusion in discrete space with uniform noise matrices
- Diffusion in discrete space via absorbing matrices, e.g., one-at-a-time masking/unmasking of individual tokens
31 items with this tag. Showing first 10 tags.
diffusion-models/protein-design
16 items with this tag. Showing first 10 tags.
diffusion-models/structure-prediction
11 items with this tag. Showing first 10 tags.
epistasis
Epistasis refers to the non-additivity of fitness effects arising from specific combinations of mutations. (1) outline three types of epistasis: magnitude epistasis (“same direction as expected but are not perfectly additive”), sign epistasis (“effect of one of the substitutions changes direction in the context of the other”), and reciprocal epistasis (“effects of both substitutions change direction when they are made together”).
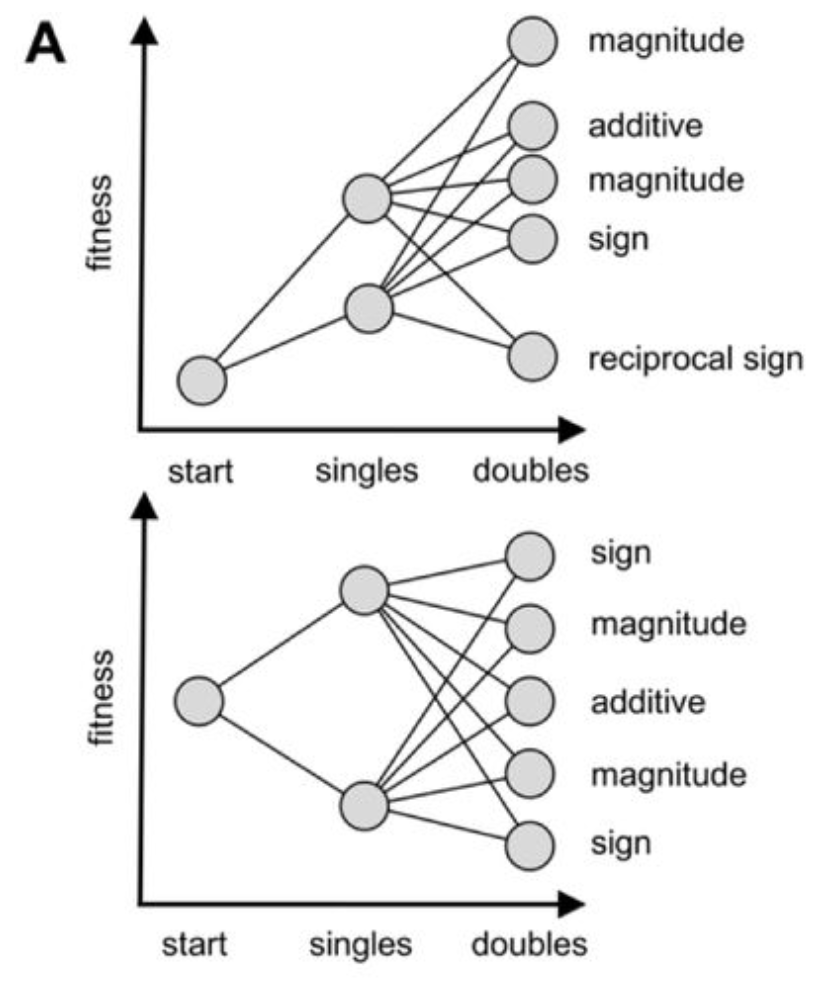 Figure from (1)
Figure from (1)
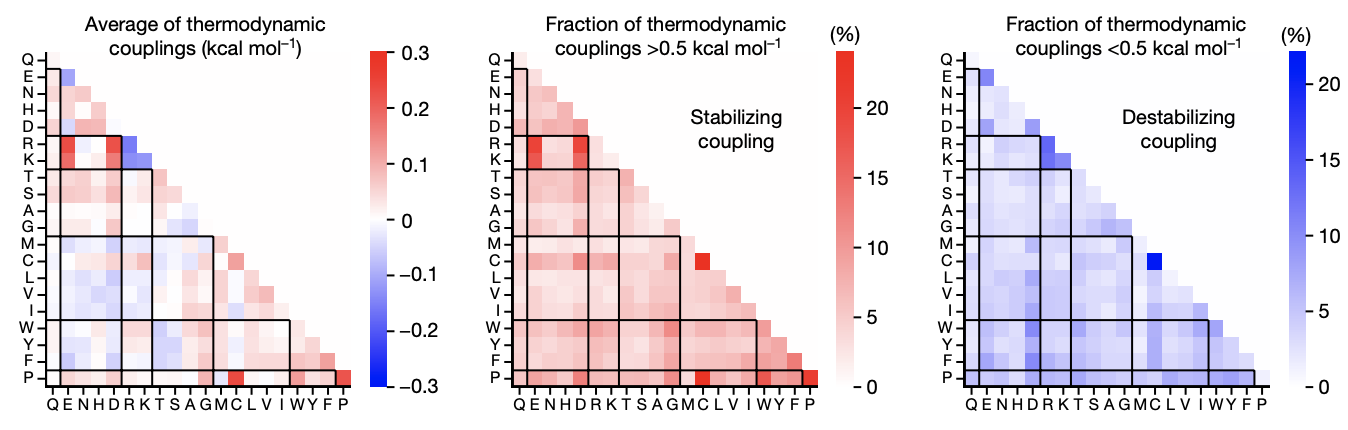 Figure from (2)
Figure from (2)
Notes
- Negative epistasis is approximately 100x more common than positive epistasis (3).
- Rates of magnitude, sign, and reciprocal epistasis are constant across positions (1). These differed from a null additive-only model with noise, which had 74% magnitude, 22% sign, and 4% reciprocal sign epistasis.
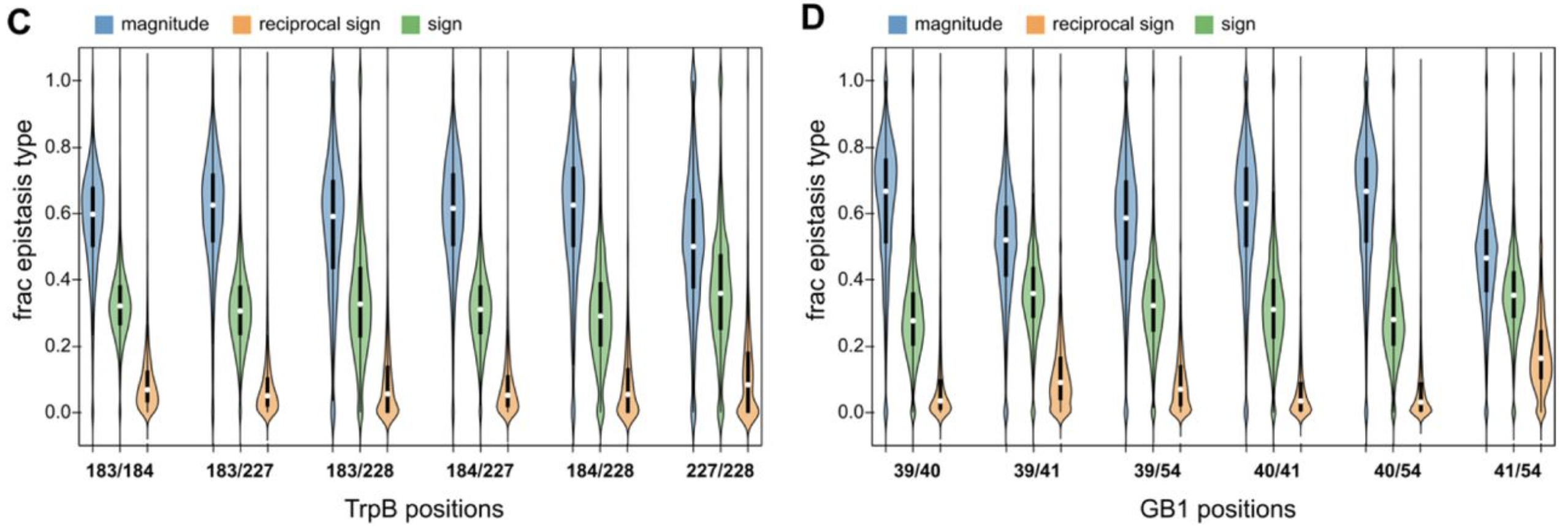 Figure from (1)
Figure from (1)
Examples
- The S373P mutation in the Spike protein is disadvantageous in pre-Omicron-variant versions of SARS-CoV-2 but advantageous in Omicron. Mentioned by Bloom and Neher (4).
- The combination of N501Y and Q498R in the Spike protein of SARS-CoV-2 increases the binding affinity to ACE2 by 387-fold. This is believed to have led to the immune-evasive mutations in the Omicron-variant and was observed in vitro by Zahradník et al. (5) prior to the emergence of Omicron:
- One example of third-order epistasis is the J-domain, where strong non-additivity is observed among a triad, but disappears if any of the three are mutated to alanine (2).
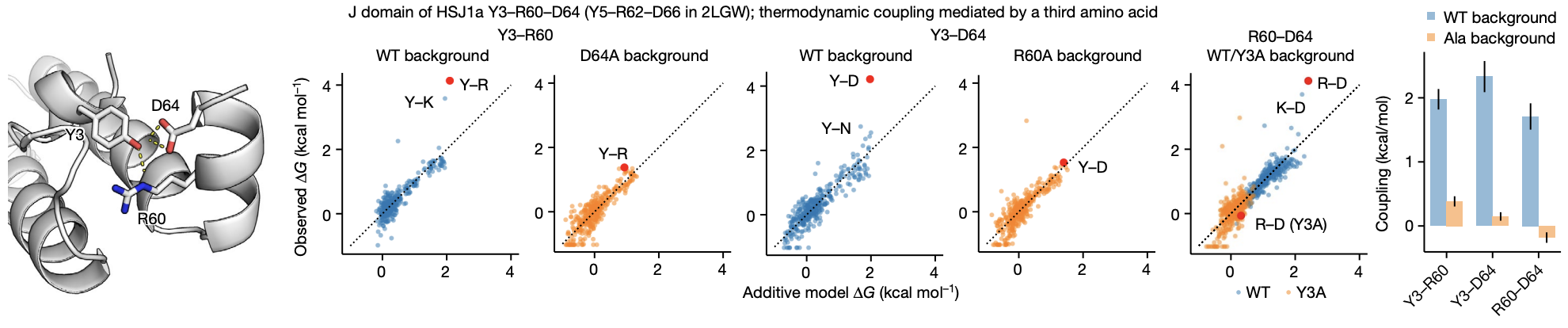 Figure from (2)
Figure from (2)
Measuring epistasis
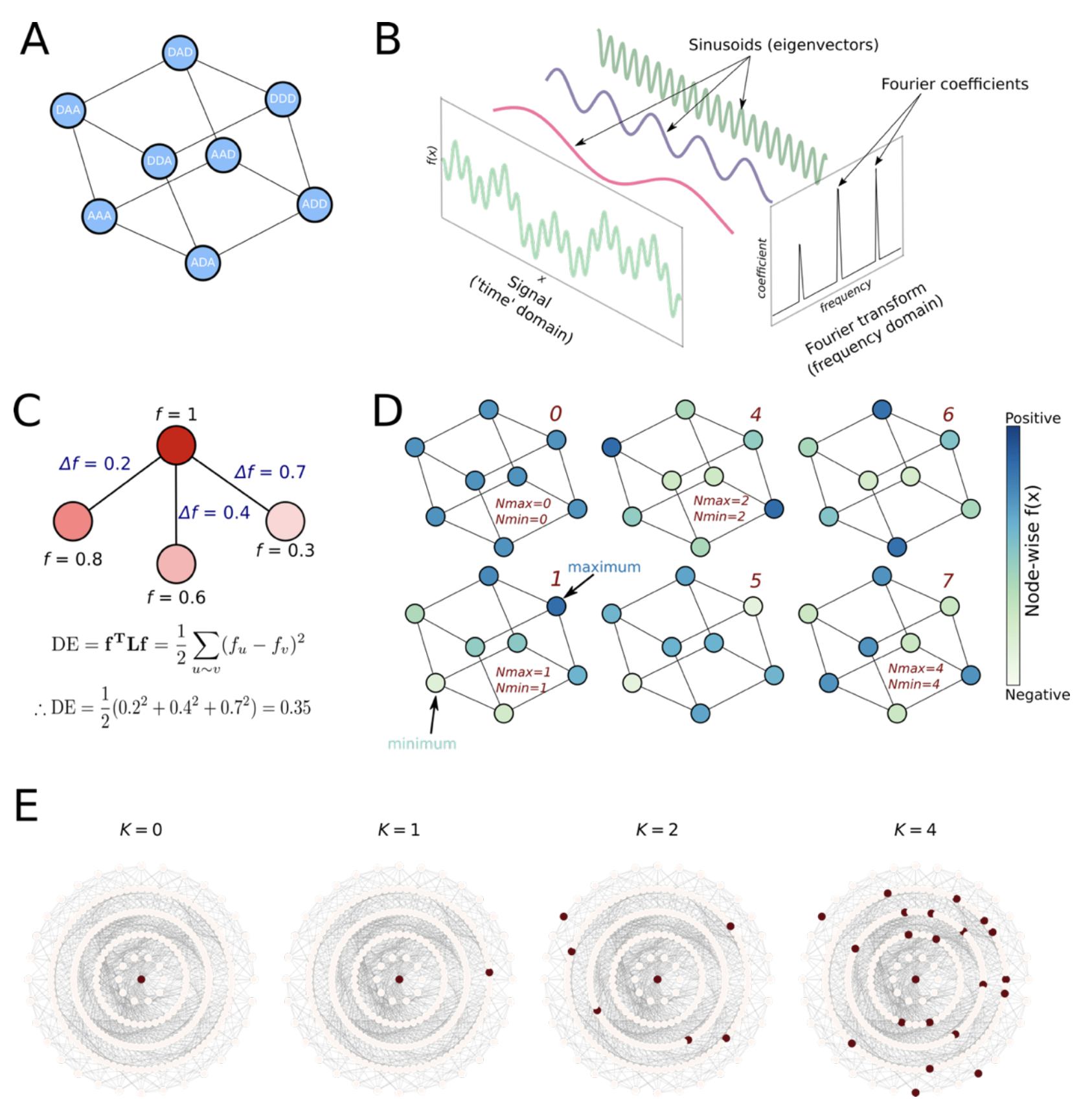 Figure from (6)
Figure from (6)
- Graph Fourier Transform: A decomposition that breaks down the signal (here, fitness) into distinct “epistatic orders”
- Dirichlet energy: quantifies how variable (i.e., rugged) the landscape is, with high energy corresponding to high ruggedness
- NK model: A linear model where is the number of sites and is the number of interacting sites; when , effect of all mutations is linear, whereas larger indicates more interactions and therefore more epistasis
12 items with this tag. Showing first 10 tags.
epitope-prediction
1 item with this tag.
evolution-and-natural-selection
Protein evolution is the process and result of gradual sequence changes resulting in functional and/or structural changes. See Epistasis for examples on why evolutionary trajectories are difficult to predict. This note excludes any discussion of somatic hypermutation.
Notes
Paradigms and preliminaries
- Neutral theory: most observed amino acid changes are neutral (i.e., silent in fitness effects). This leads to genetic drift. Developed by Kimura (1).
- Nearly neutral theory: deleterious mutations are retained and subsequently compensated for by advantageous mutations (which is consistent with the observation that most missense mutations are destabilizing). Developed by Ohta (2) to explain why the rate of protein evolution was independent of generation time, which is in turn inversely proportional to population size.
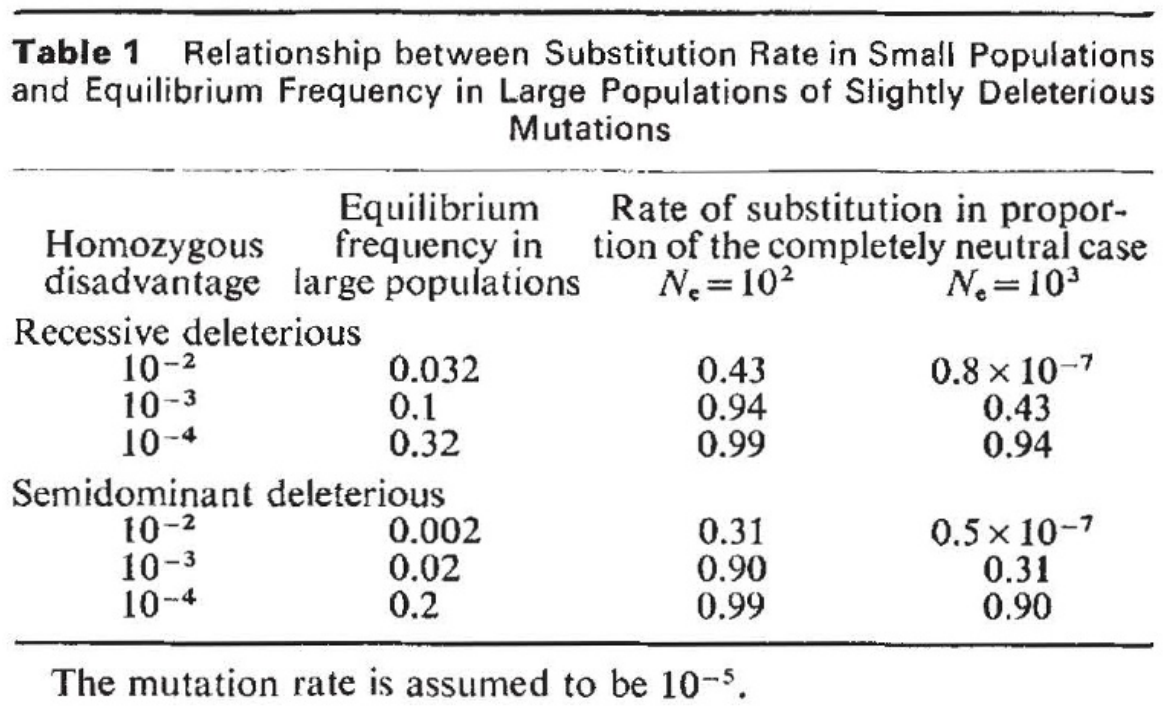 Figure from (2)
Figure from (2) - The theory of punctuated equilibrium suggests that phenotypes change very little for long stretches of time, followed by abrupt rapid changes (3).
- Statistical physics approach: population size is equated with inverse temperature (such that infinite population is analogous to zero degrees Kelvin), and log-fitness with energy (4). Advantageous and deleterious mutations are predicted to occur with equal frequency. This framing ignores the imbalance in sequence data (5,6).
- Fisher’s geometric model: The overall fitness of a phenotype can be quantified along dimensions; Fisher postulated that phenotypes in a population were distributed as a hypersphere centered on a local maximum.
- Protein evolvability refers to the ability of a protein to 1) evolve new functions in relatively few mutations and 2) be robust to mutations that lead to loss-of-function (7). These are described as contradictory statements by Tokuriki & Tawfik (8) but are described as complementary at the structural level.
- “The principle of minimal frustration suggests that naturally evolved proteins with the same structure should have similar folding rates and that modulation of thermodynamic stability should occur via unfolding rates” (quoted from (9)). This has been supported by the observation that thioredoxins fold at similar rates but unfold at rates that correlate with their thermostability values.
Observations
- Protein folds with high sequence diversity also have high functional diversity (7).
- The sequence capacity of a protein exceeds for even small proteins (35-40 AAs), but the fraction of stable states is extremely small and inversely correlated with protein size. These values were estimated using Potts models (10).
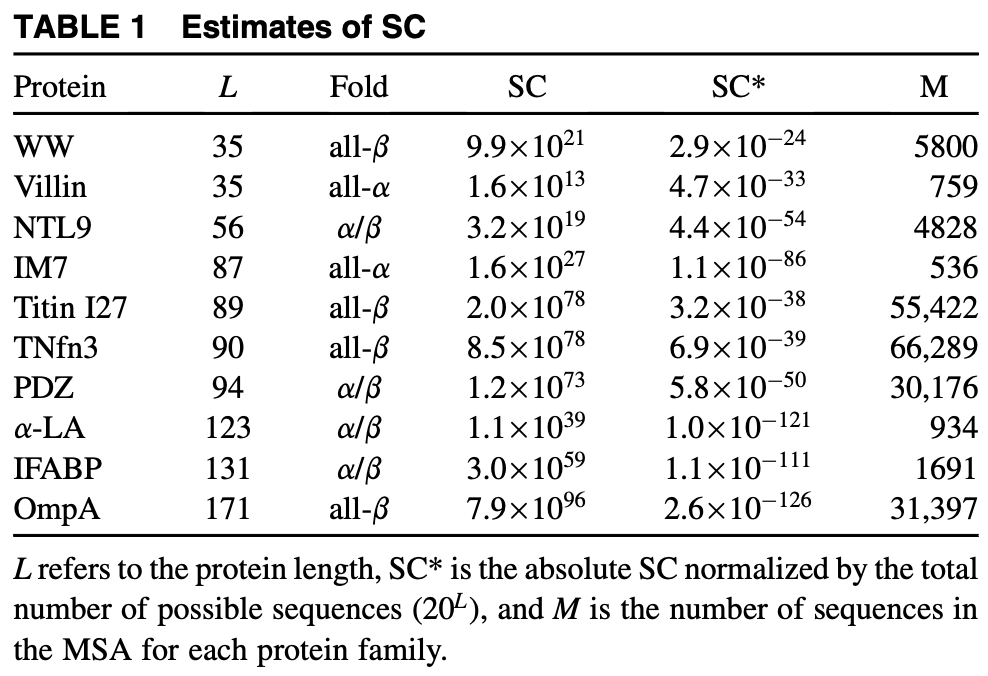 Figure from (10)
Figure from (10)
23 items with this tag. Showing first 10 tags.
evolution-and-natural-selection/structure
5 items with this tag.
immune-repertoires
Immune repertoires are the full breadth of B-cell and T-cell receptors being expressed by a human that are available for potential antigen binding. Rees (1) provides estimates suggesting that humans have naive repertoires of about sequences.
Related:
5 items with this tag.
inverse-folding
Inverse folding describes the problem of designing a sequence for a structure. Typically these are limited to the twenty canonical amino acids.
Methods
See Hybrid sequence-structure models for a list of methods that incorporate PLMs
- ProteinMPNN and its derivatives
- ESM-IF
- GearNet
- Frame2seq
- GVP-GNN
Notes
Training
- Training inverse folding models with backbone dihedral angles as features usually improved sequence recovery (1).
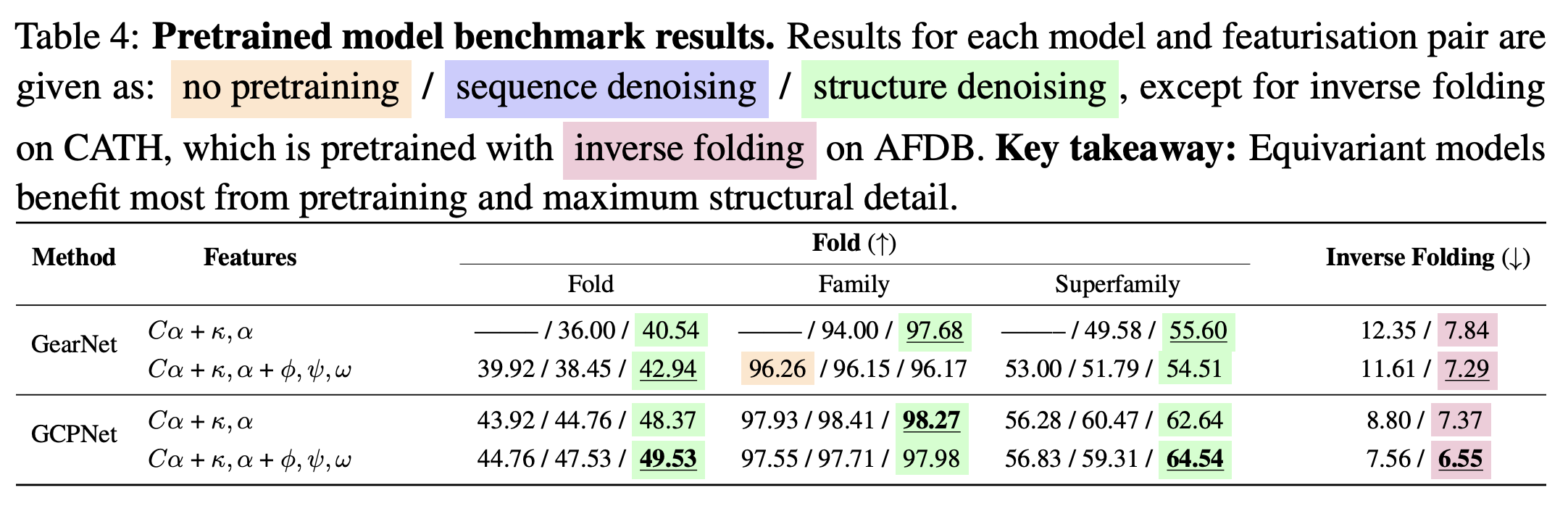 Figure from (1)
Figure from (1)
Execution
- Forward-folding is a stronger predictor of inverse folding success than sequence recovery ((2), citing Watson et al. (3,4)).
Datasets
- PDBench is a dataset of 595 protein structures with diverse, evenly divided topologies for benchmarking of Inverse folding methods (5).
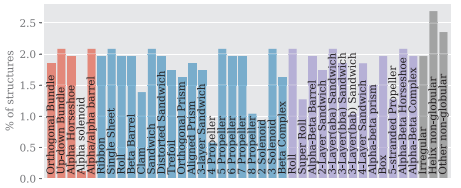 Figure 2 from Castorina et al. (5)
Figure 2 from Castorina et al. (5)
48 items with this tag. Showing first 10 tags.
inverse-folding/evaluation
35 items with this tag. Showing first 10 tags.
inverse-folding/training
10 items with this tag.
ligand-docking
7 items with this tag.
light-chains
The light chain of an antibody makes up part of its variable region and Fab, and therefore is involved in antigen binding. In humans, the Kappa and Lambda subtypes are found and split about 60:40, whereas in mice it is closer to 90:10.
Kappa and lambda subtype
- The ratio of kappa to lambda in circulating antibodies is about 60:40; when this falls out of balance, that can be a symptom of B cell lymphoma.
- Lambda light chains are more flexible than kappa light chains due to an extra glycine in the switch region (_Articles that need citations).
6 items with this tag.
low-rank-adaptation
7 items with this tag.
machine-learning
2 items with this tag.
pae
Predicted aligned error (PAE) is a measurement calculated by protein structure prediction neural networks to capture positional errors between two amino acids in a computational model. It was introduced by AlphaFold2. A derivative metric, ipSAE, has been shown to be more robust at identifying potential binders.
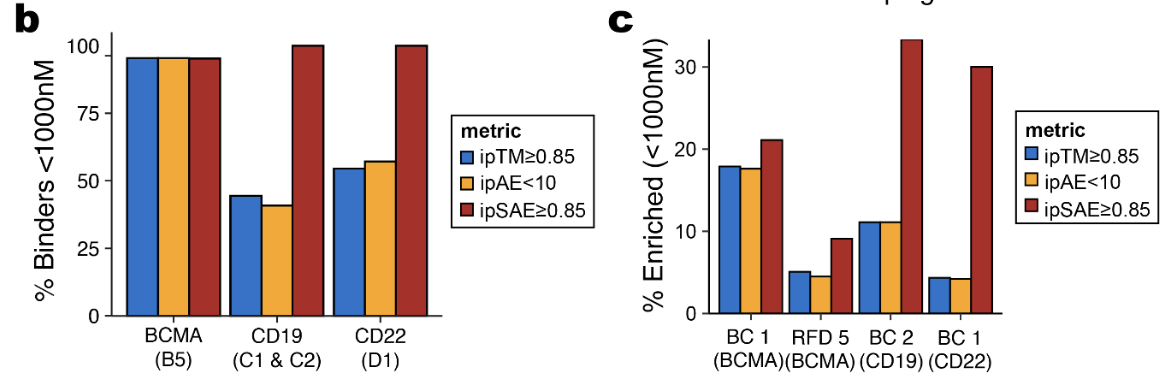 Figure from (1)
Figure from (1)
9 items with this tag.
plddt
(LDDT redirects here) pLDDT (predicted local distance difference test) is a confidence metric used by neural networks for protein structure prediction. It captures the per-residue accuracy, both in terms of neighborhood and side chain rotamer. It was first directly integrated into structure prediction by AlphaFold2 at the per-residue level and has been widely adopted since. AlphaFold3 adopted per-atom pLDDT.
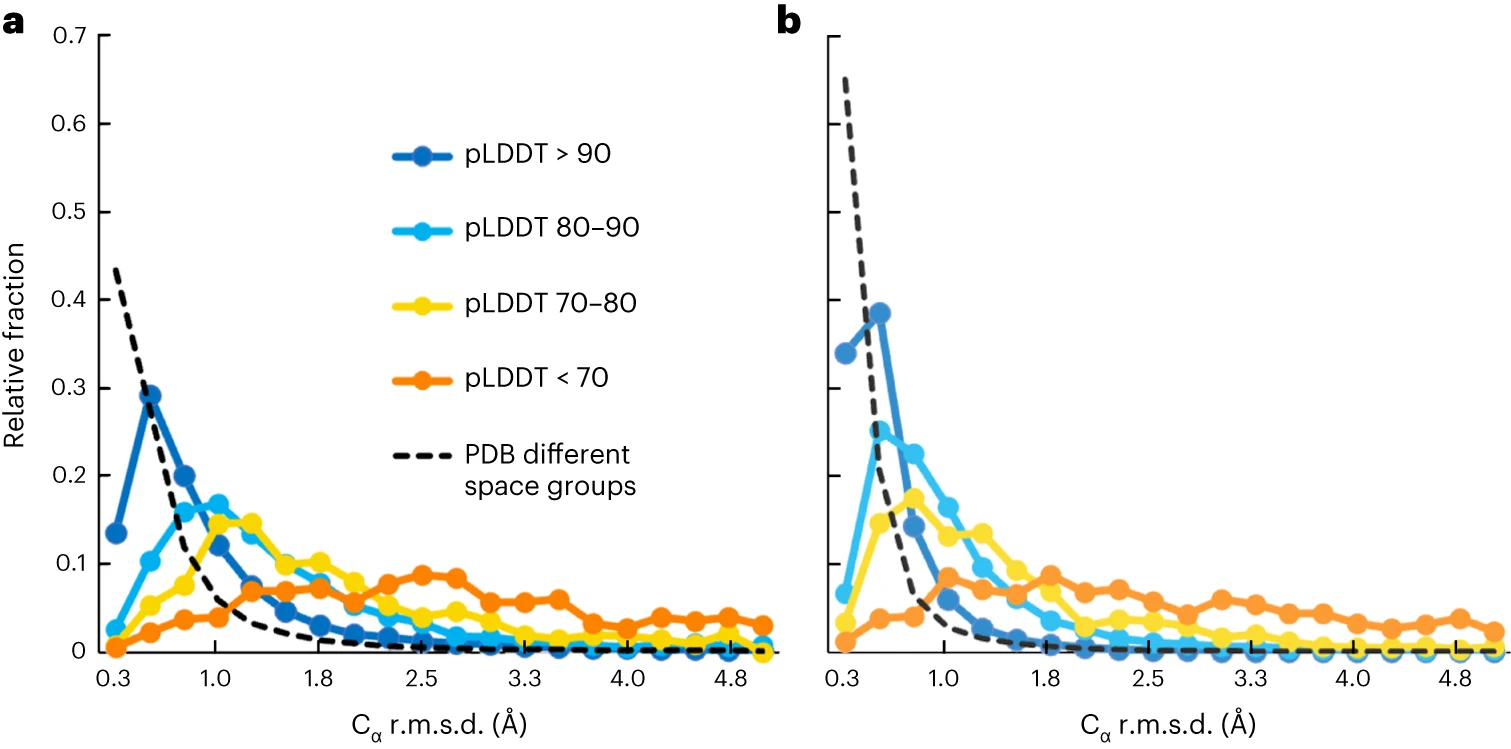 Figure from (1)
Figure from (1)
Notes
- When clustering predicted protein structures, sparse clusters tend to have lower pLDDT (2). This was found to be independent of MSA depth.
- pLDDT correlates poorly with GDT-TS among AlphaFold2 models in CASP15. This was observed in a repeat that used deeper MSAs (3).
- De novo sequences designed by inversion with high pLDDT were found by ESM to have high perplexity (4).
- While the default pLDDT is not continuously differentiable and thus unsuitable for training, (5) use a modified version that can be used as a loss function.
- pLDDT can be used as spatial restraints in biomolecular simulations: The equation was originally presented by Hiranuma et al. (6) and was used by del Alamo et al. (7) as coordinate constraints in Rosetta when refining AlphaFold2 models.
28 items with this tag. Showing first 10 tags.
protein-backbone-design
Protein backbone design is the generation of protein backbones in three-dimensional space. This section also covers generation and design of entire protein structures in Cartesian space, but most methods uncouple design of the backbone and design of the sequence given the backbone (inverse folding). As of May 2024, the current state of the art uses diffusion.
Methods
- Chroma (1)
- RF-diffusion (2) and RFam (3)
- Hallucination using AlphaFold2 and RosettaFold
- Inpainting using RosettaFold (4)
Datasets
- Verkuil et al. (5) use a test set of 39 PDBs for their validation, although they cite someone else:
- 1QYS
- 2KL8
- 2KPO
- 2LN3
- 2LTA
- 2LVB
- 2N2T
- 2N2U
- 2N3Z
- 2N76
- 4KY3
- 4KYZ
- 5CW9
- 5KPE
- 5KPH
- 5L33
- 5TPJ
- 5TRV
- 6CZG
- 6CZH
- 6CZI
- 6CZJ
- 6D0T
- 6DG6
- 6DKM A
- 6DKM B
- 6DLM A
- 6DLM B
- 6E5C
- 6LLQ
- 6MRR
- 6MRS
- 6MSP
- 6NUK
- 6W3F
- 6W3W
- 6WI5
- 6WVS
- 7MCD
24 items with this tag. Showing first 10 tags.
protein-backbone-design/designability
22 items with this tag. Showing first 10 tags.
protein-design
Auto-generated
This page is generated automatically from notes tagged
protein-design/*. Add prose above the generated marker to preserve it across regenerations.
33 items with this tag. Showing first 10 tags.
protein-design/design
21 items with this tag. Showing first 10 tags.
protein-design/enzyme-design
6 items with this tag.
protein-folding
Not to be confused with protein structure prediction
Protein folding is the process by which an amino acid polypeptide self-organizes into a 3D structure.
Prediction
26 items with this tag. Showing first 10 tags.
protein-folding/structure-prediction
10 items with this tag.
protein-folding/unfolding
16 items with this tag. Showing first 10 tags.
protein-language-models
Protein language models (PLMs) are a type of Transformer model trained on either protein sequences or Multiple sequence alignments.
Methods
Single-sequence
- ESM: currently the most widely-used encoder PLM
- ProGen: probably the most widely-used decoder PLM
- ProtBERT and DistillProtBERT (1)
- ProteinNPT
- xTrimoPGLM
- CARP: A CNN that performs as well as transformer-based methods on both pretraining and downstream tasks. Anecdotally, these can’t indirectly calculate contact maps via the Categorical Jacobian method as well as transformer-based models.
- DASM (deep amino acid sequence model), which is trained on germline-descendant point mutation pairs to learn relative mutation frequencies, after normalizing for expected mutation frequencies in the codon table (2).
Notes
General observations
- PLMs are in-context learners that default to retrieving information from nearby repeats (3).
Representations
- Multiple instance learning using PLM embeddings of all genes in a viral genome identifies which sequences are responsible for host tropism (4). For example, this ranked the Spike protein as the key contributor of host tropism.
- Homolog detection using PLM representations can be improved by compression (5). Using the full representations worsened detection AUC by 7.4%.
- PLMs with a smoother representation space are better predictors of protein function (6).
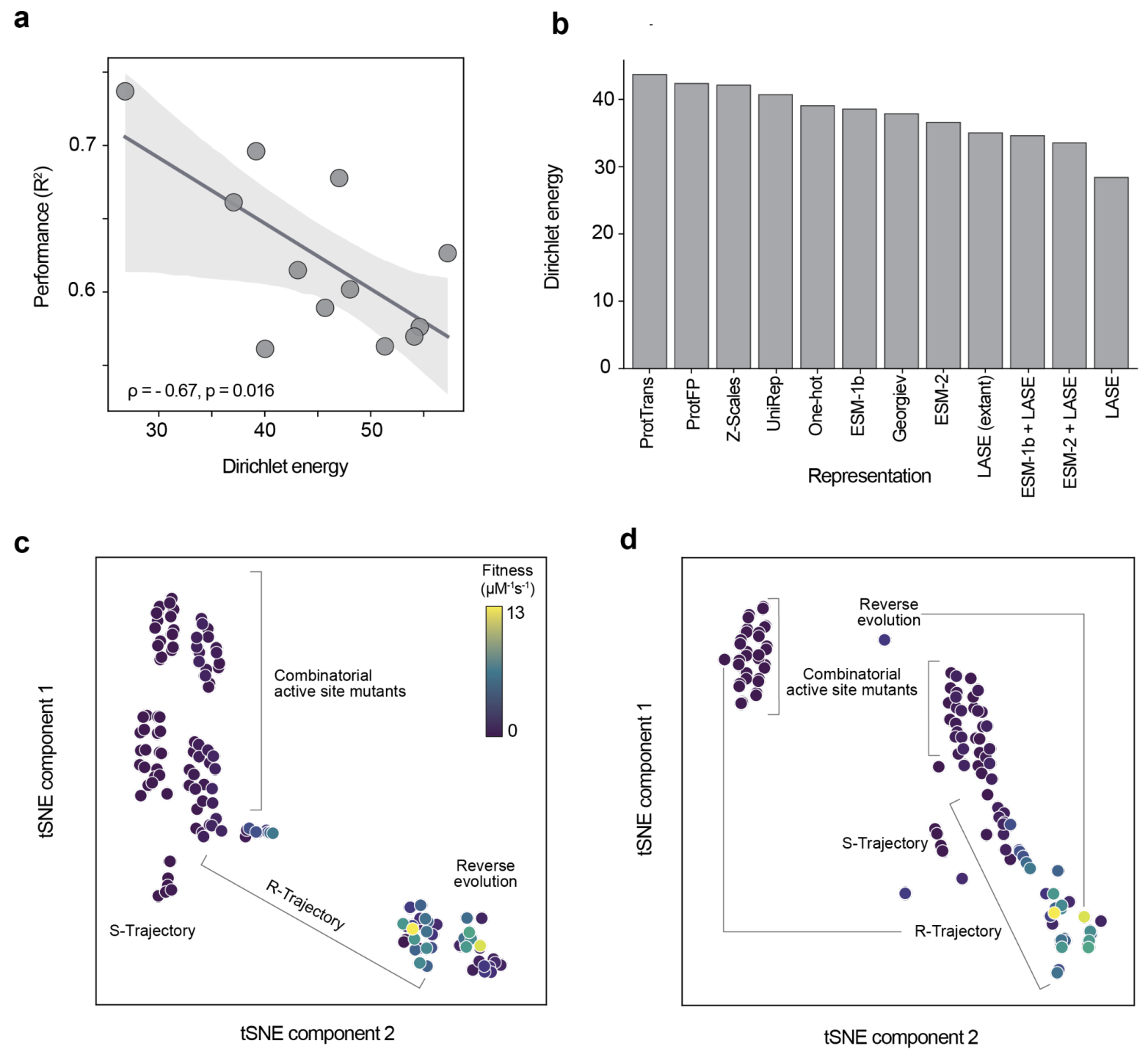 Figure from (6)
Figure from (6)
Hybrid PLM-inverse folding models
From Hybrid sequence-structure models
Training
- Matthews et al. (6) found that masking 0.5% of residues when training PLMs improved predictive performance (greater ) relative to 15% used by ESM.
104 items with this tag. Showing first 10 tags.
protein-language-models/antibodies
24 items with this tag. Showing first 10 tags.
protein-language-models/representations
42 items with this tag. Showing first 10 tags.
protein-language-models/training
32 items with this tag. Showing first 10 tags.
protein-protein-interactions
Protein-protein interactions describe when two or more proteins bind to one another.
21 items with this tag. Showing first 10 tags.
protein-structure-tokenization
Protein structure tokenization refers to the process of discretizing protein structure using a learned codebook derived from vector-quantized variational autoencoders. It was first used for search purposes with Foldseek and has since been adopted for use with ESM3, SaProt, and others.
Notes
- Structural tokenization circumvents the inability of inverse folding models to be effectively trained on exclusively computational models.
rosettafold
RosettaFold (sometimes stylized as RoseTTAfold) is a protein structure prediction method unveiled in mid-2021 (1). A second version, RosettaFold2, was released in early 2023 (2). Its architecture closely tracks that of AlphaFold2, with several changes, such as the use of an SE3-transformer.
1 item with this tag.
structure-prediction
Structure prediction refers to the problem of predicting the 3D shape of a protein or nucleotide sequence without any experimental information. Common metrics used for evaluating the quality of predicted structures include LDDT (residue-level, TM-score (whole-structure level), and DockQ (complex level).
Methods
MSA-based
- AlphaFold2: currently viewed as the highest-accuracy method
- RosettaFold
- Diffold: A fine-tuned version of AlphaFold2
PLM-based
- ESMFold: currently the most widely-used method, albeit probably not the most accurate model in this category
- OmegaFold
- xTrimoPGLM
Others
- EquiFold: a method that needs to be fine-tuned on specific families of proteins
- EigenFold: a method that uses diffusion to model the dynamics of proteins, albeit unsuccessfully
For antibodies
See Antibody structure prediction
Notes
Training
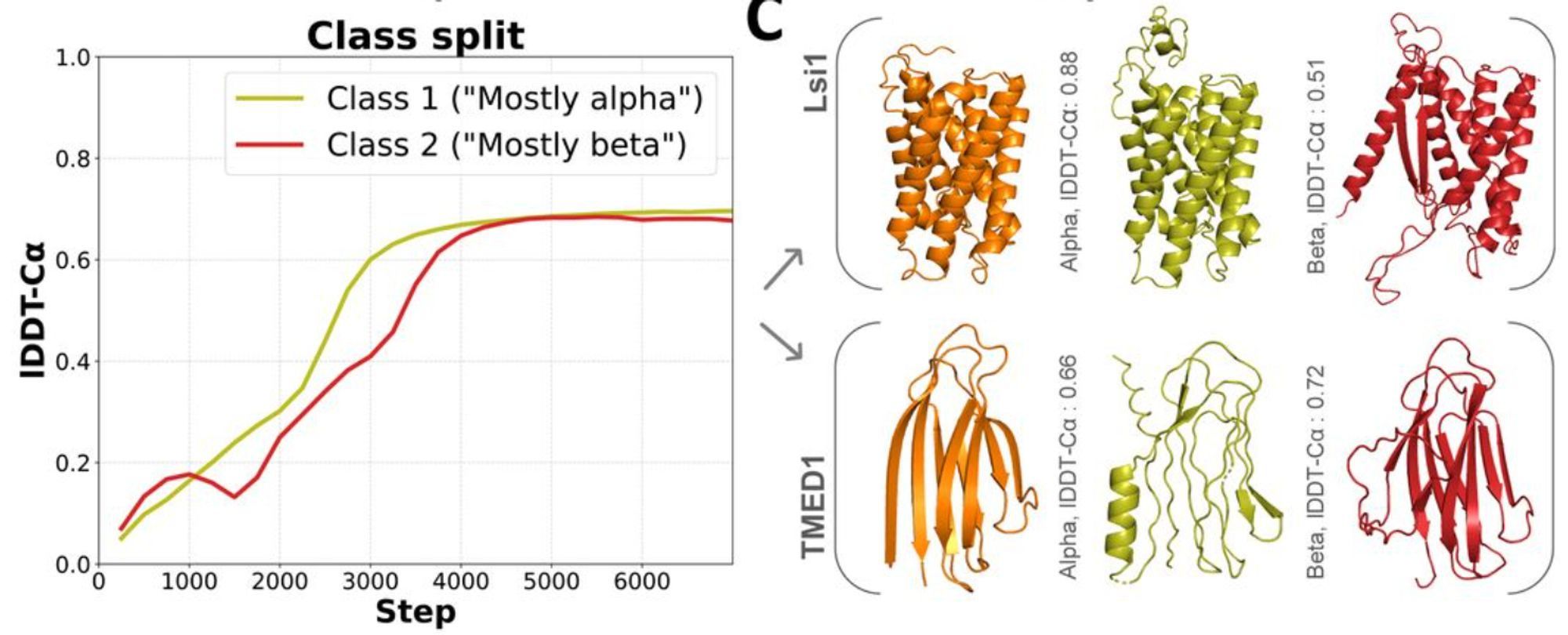 Figure from (1)
Figure from (1)
Sidechain prediction
- Formulating the sidechain prediction problem as a classification problem by binning chi angles, rather than a regression problem, let to improved performance (2).
- Sidechain prediction methods not sensitive to B-factor cutoffs. The outcome of sidechain prediction model PIPPack was not strongly affected by B-factor values of protein structures in the training set (2).
88 items with this tag. Showing first 10 tags.
structure-prediction/architecture
9 items with this tag.
structure-prediction/complex-prediction
8 items with this tag.
structure-prediction/limitations
20 items with this tag. Showing first 10 tags.
structure-prediction/metrics
9 items with this tag.
structure-prediction/sampling
27 items with this tag. Showing first 10 tags.
structure-prediction/training
13 items with this tag. Showing first 10 tags.
thermostability
Thermostability refers to a protein’s ability to remain folded at high temperatures or under harsh conditions. It is a highly desirable property for engineered proteins.
Prediction
- Phantom epistasis refers to the inclusion of unnecessary model parameters when building biophysical/statistical fitness models (Fitness prediction). Faure et al. (1) attribute this to the epistasis mechanisms reviewed by Domingo et al. (2).
- Thermodynamic reversability can be used for expanding training sets for stability prediction/ddG prediction ML models. However, it has been shown to lead to biases that favor WT amino acids. Diaz et al. (3) claim to mitigate this.
- The amount of ddG data available for a given residue for training can be expanded using thermodynamic permutation, where measurements are increased to . This was used by MutComputeXGT on the Tsuboyama et al. (4) dataset. It is useful for stability prediction and improves generalization in (3).
- ddG data is skewed with hydrophobic amino acids (e.g., alanine scans). This has been reported to increase solvation ddG by 0.8 kcal/mol in studies cited by (3). The Tsuboyama et al. (4) data does not have this bias.
58 items with this tag. Showing first 10 tags.
thermostability/design
7 items with this tag.
thermostability/determinants
6 items with this tag.
thermostability/evolution
5 items with this tag.
thermostability/mutations
7 items with this tag.
thermostability/prediction
23 items with this tag. Showing first 10 tags.
tm-score
Summary
TM-score is an alignment-dependent protein structure similarity term introduced by (1) that is widely used for assessing protein structure prediction methods. It is defined as:
: length of the amino acid sequence of the target protein : number of residues in both the target and query proteins : Distance between pairs of residues : Distance scaling factor
Predicted variants
Protein folding neural networks such as AlphaFold2 and AlphaFold3 predict a distribution over aligned errors for each ordered residue pair, then use that distribution to calculate pTM and ipTM confidence scores (2,3). For bin center and probability , the expected TM contribution for a residue pair is:
ipTM (interface predicted TM-score) uses this same TM-score transform, but masks the average to residues in other chains:
is the number of modeled residues used for . In practice, ipTM is an inter-chain pTM score rather than a direct interface-contact score: all residues in other chains can contribute, not just residues close in 3D.
ipSAE (interaction prediction score from aligned errors) is a PAE-derived replacement for ipTM calculated after choosing a PAE cutoff (4). For a chain direction and aligned residue , it keeps only residues in the other chain with and computes:
Then:
The reported pairwise “max” score is the maximum of the two chain directions. Compared with ipTM, ipSAE specifically improves robustness to construct length, disordered tails, and accessory domains because it scores only confident inter-chain residue pairs, uses a local based on those residues rather than full chain/complex length, and uses the output PAE matrix rather than requiring AlphaFold’s internal aligned-error logits (4).
20 items with this tag. Showing first 10 tags.
transformers
7 items with this tag.
variant-effect-prediction
Variant effect prediction covers the changes in properties or fitness (measured in various ways) resulting from small sequence-level changes in proteins.
25 items with this tag. Showing first 10 tags.